

Classic infantile-onset Pompe disease with histopathological neurologic findings linked to a novel GAA gene 4 bp deletion: A case study
- PMID: 35532199
- PMCID: PMC9266604
- DOI: 10.1002/mgg3.1957
Classic infantile-onset Pompe disease with histopathological neurologic findings linked to a novel GAA gene 4 bp deletion: A case study
Abstract
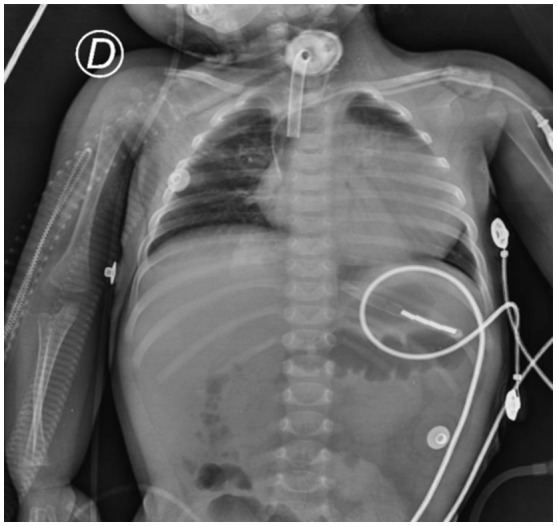
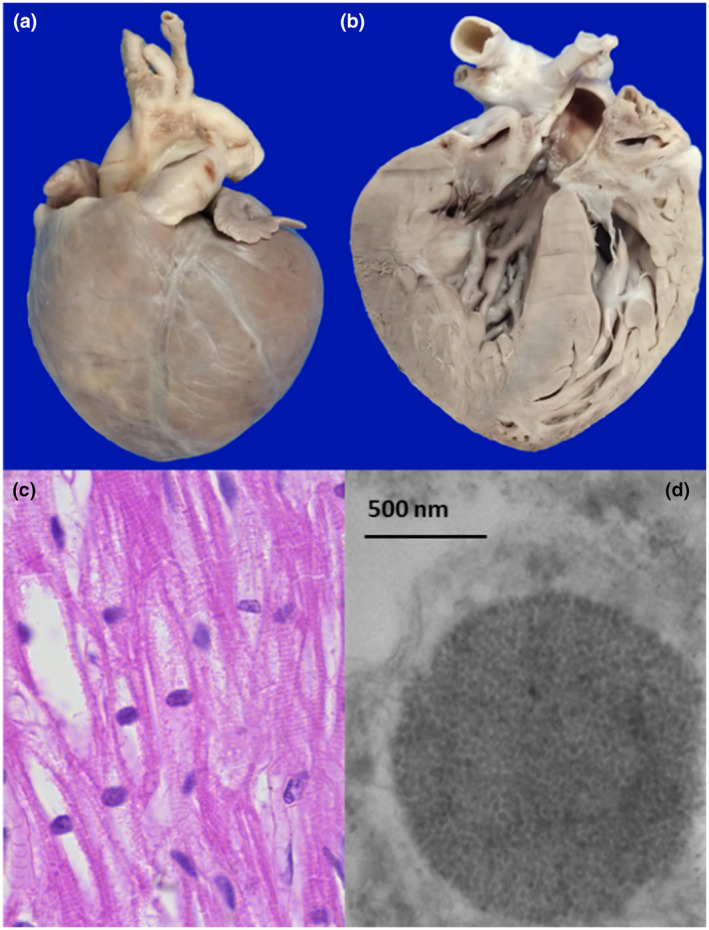
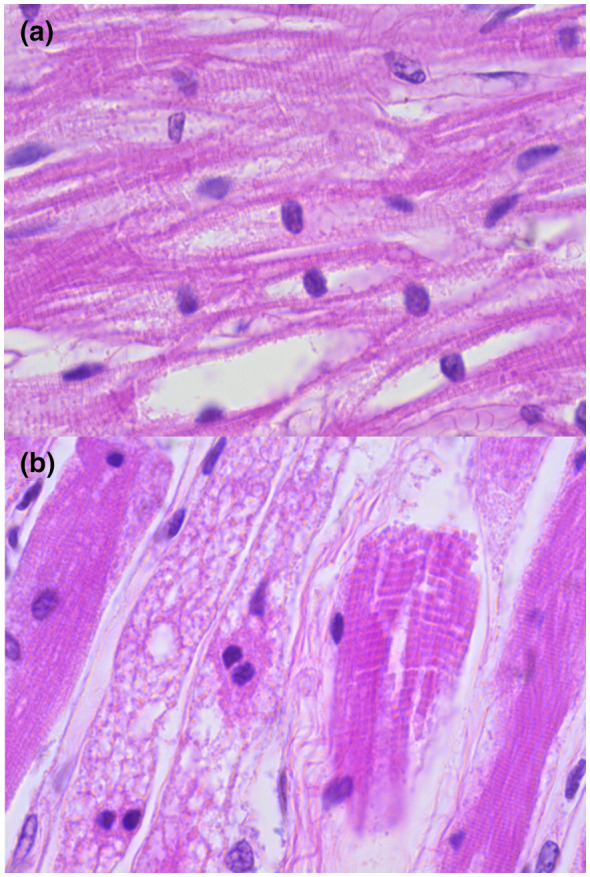
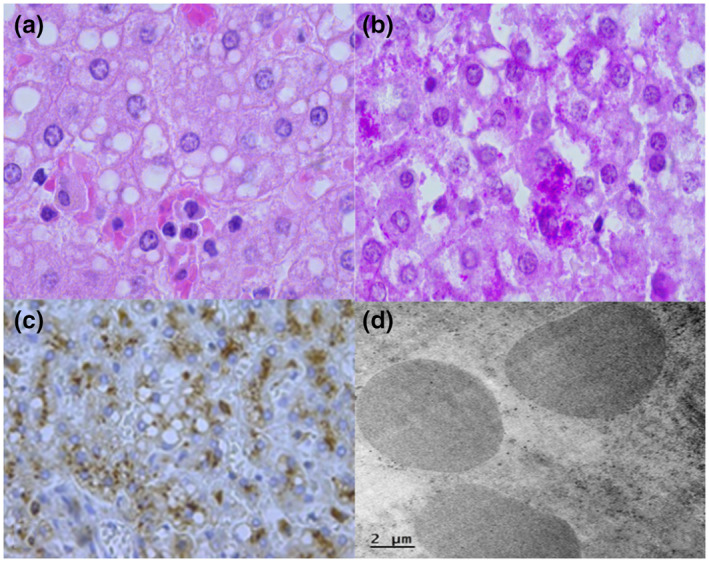
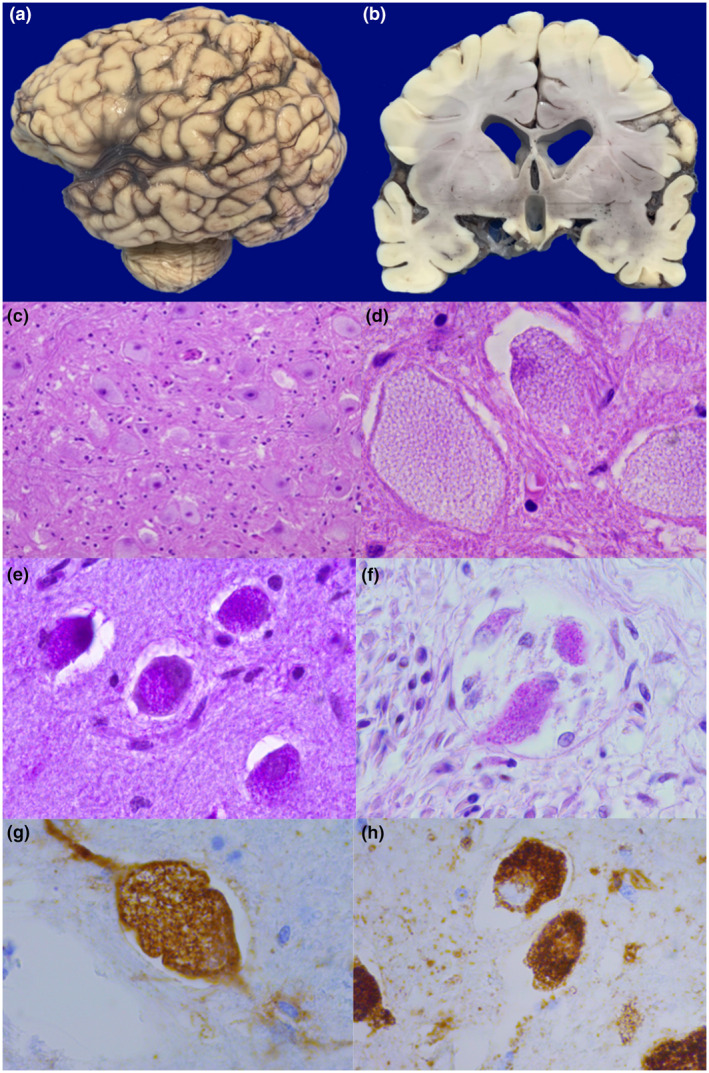
Pompe disease (PD) is an autosomal recessive disorder by a deficiency of acid α-glucosidase (GAA) with intralysosomal glycogen accumulation in multiple tissues. We present the case of a 5-month-old male with hypertrophic cardiomyopathy, hypotony, feeding difficulties, and oxygen requirement since birth. At 3 months of age, he develops heart failure, respiratory impairment, and neurological deterioration. The echocardiogram revealed concentric hypertrophic cardiomyopathy with left-diastolic dysfunction. We found increased creatine-phosphokinase, lactate dehydrogenase, and urinary glucose tetrasaccharide levels, 50% of PAS-positive vacuolated lymphocytes in the peripheral blood smear, and low GAA activity. Sequencing of coding exons and flanking intronic sequences revealed a novel homozygous 4 bp deletion in exon 15 of the GAA gene (c.2066_2069delAGCC/p.Glu689Glyfs*6). IOPD was diagnosed. At 5 months old, we started enzyme replacement therapy with an alpha-alglucosidase of 20 mg/kg weekly and immunomodulation with intravenous immunoglobulin. He developed two cardiorespiratory arrests with subsequent neurologic deterioration, convulsive crisis, and respiratory failure and died at 9 months old. We found the usual PD hallmarks in the heart, striated muscle, and liver but also we found neuronal lesions characterized by cytoplasm vacuolization with PAS-positive granules in the central nervous system and myenteric plexus. We describe a novel GAA gene pathogenic variant with a particular phenotype characterized by classic IOPD and neurologic histopathological findings. Enhancing the knowledge of lysosomal diseases is critical to improving the diagnosis and treatment of these patients.
Keywords: GAA gene; acid alpha-glucosidase; c.2066_2069delAGCC; infantile-onset Pompe disease; neurologic; p.Glu689Glyfs*6.
© 2022 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals LLC.
Conflict of interest statement
Magdalena Cerón‐Rodríguez, Pedro Valencia‐Mayoral, and Argelia Escobar Sánchez have received a fee for speaking from different pharmaceutical companies. Juan‐Luis Salgado‐Loza has received payment for writing assistance from pharmaceutical companies. Daniela Castillo‐García, Carlos Patricio Acosta‐Rodríguez‐Bueno, Jesús Aguirre‐Hernández, Juan Rafael Murillo Eliosa do not have any conflict of interest to declare. The authors confirm independence from pharmaceutical companies. The information of the article has not been influenced by pharmaceutical companies.
Figures
Similar articles
-
Genotype, phenotype and treatment outcomes of 17 Malaysian patients with infantile-onset Pompe disease and the identification of 3 novel GAA variants.Orphanet J Rare Dis. 2023 Aug 4;18(1):231. doi: 10.1186/s13023-023-02848-6. Orphanet J Rare Dis. 2023. PMID: 37542277 Free PMC article.
-
CRISPR-mediated generation and characterization of a Gaa homozygous c.1935C>A (p.D645E) Pompe disease knock-in mouse model recapitulating human infantile onset-Pompe disease.Sci Rep. 2022 Dec 14;12(1):21576. doi: 10.1038/s41598-022-25914-8. Sci Rep. 2022. PMID: 36517654 Free PMC article.
-
Clinical course, mutations and its functional characteristics of infantile-onset Pompe disease in Thailand.BMC Med Genet. 2019 Sep 11;20(1):156. doi: 10.1186/s12881-019-0878-8. BMC Med Genet. 2019. PMID: 31510962 Free PMC article.
-
Gene Therapy for Pompe Disease: The Time is now.Hum Gene Ther. 2019 Oct;30(10):1245-1262. doi: 10.1089/hum.2019.109. Epub 2019 Sep 9. Hum Gene Ther. 2019. PMID: 31298581 Review.
-
Pompe disease: early diagnosis and early treatment make a difference.Pediatr Neonatol. 2013 Aug;54(4):219-27. doi: 10.1016/j.pedneo.2013.03.009. Epub 2013 Apr 28. Pediatr Neonatol. 2013. PMID: 23632029 Review.
Cited by
-
Neurological glycogen storage diseases and emerging therapeutics.Neurotherapeutics. 2024 Sep;21(5):e00446. doi: 10.1016/j.neurot.2024.e00446. Epub 2024 Sep 14. Neurotherapeutics. 2024. PMID: 39277505 Free PMC article. Review.
-
Highlights of Precision Medicine, Genetics, Epigenetics and Artificial Intelligence in Pompe Disease.Int J Mol Sci. 2025 Jan 17;26(2):757. doi: 10.3390/ijms26020757. Int J Mol Sci. 2025. PMID: 39859472 Free PMC article. Review.
-
Severe CNS involvement in a subset of long-term treated children with infantile-onset Pompe disease.Mol Genet Metab. 2024 Feb;141(2):108119. doi: 10.1016/j.ymgme.2023.108119. Epub 2023 Dec 22. Mol Genet Metab. 2024. PMID: 38184429 Free PMC article.
References
-
- Banugaria, S. G. , Prater, S. N. , Patel, T. T. , DeArmey, S. M. , Milleson, C. , Sheets, K. B. , Bali, D. S. , Rehder, C. W. , Raiman, J. A. J. , Wang, R. A. , Labarthe, F. , Charrow, J. , Harmatz, P. , Chakraborty, P. , Rosenberg, A. S. , & Kishnani, P. S. (2013). Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material‐negative classic infantile Pompe disease: A step towards improving the efficacy of ERT. PLoS ONE, 8, e67052. 10.1371/journal.pone.0067052 - DOI - PMC - PubMed
-
- Becker, J. A. , Vlach, J. , Raben, N. , Nagaraju, K. , Adams, E. M. , Hermans, M. M. , Reuser, A. J. J. , Brooks, S. S. , Tifft, C. J. , Hirschhorn, R. , Huie, M. L. , Nicolino, M. , & Plotz, P. H. (1998). The African origin of the common mutation in African American patients with glycogen‐storage disease type II [3]. American Journal of Human Genetics, 62, 991–994. 10.1086/301788 - DOI - PMC - PubMed
-
- Burt, A. , Ferrell, L. , & Hubscher, S. (2017). Chapter 3. Developmental and inherited liver disease. MacSween's pathology of the liver (7th ed.). Elsevier. https://www.eu.elsevierhealth.com/macsweens‐pathology‐of‐the‐liver‐97807...
-
- Byrne, B. J. , Fuller, D. D. , Smith, B. K. , Clement, N. , Coleman, K. , Cleaver, B. , Vaught, L. , Falk, D. J. , McCall, A. , & Corti, M. (2019). Pompe disease gene therapy: Neural manifestations require consideration of CNS directed therapy. Annals of Translational Medicine, 7(13), 290. 10.21037/atm.2019.05.56 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
