

Dysbiosis in the Gut Microbiota in Patients with Inflammatory Bowel Disease during Remission
- PMID: 35532243
- PMCID: PMC9241752
- DOI: 10.1128/spectrum.00616-22
Dysbiosis in the Gut Microbiota in Patients with Inflammatory Bowel Disease during Remission
Abstract
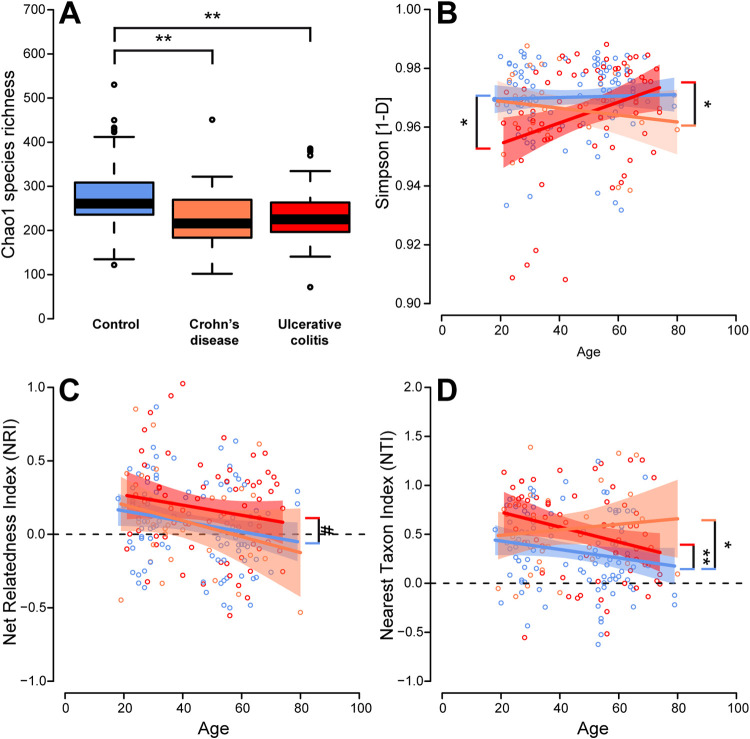
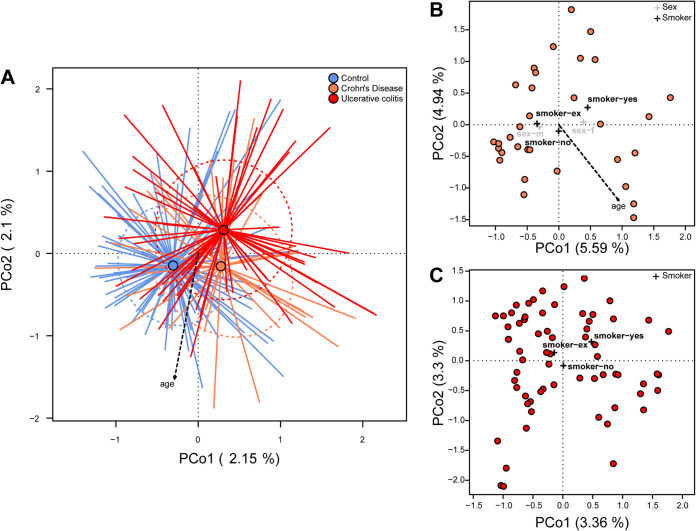
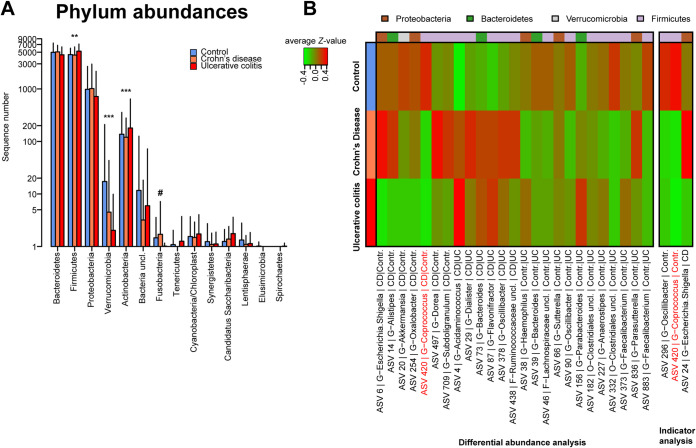
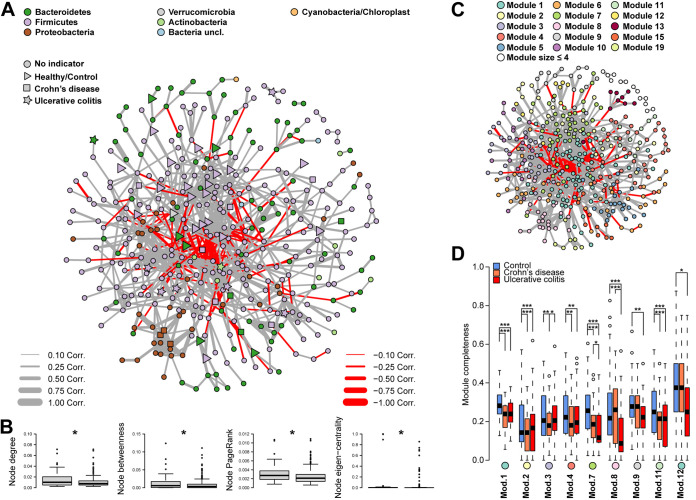
Inflammatory bowel disease (IBD) is a chronic, relapsing, inflammatory disorder which comprises two main conditions: Crohn's disease (CD) and ulcerative colitis (UC). Although the etiology of IBD has not been fully elucidated, the gut microbiota is hypothesized to play a vital role in its development. The aim of this cross-sectional study was to characterize the fecal microbiota in CD or UC patients in a state of remission to reveal potential factors sustaining residual levels of inflammation and triggering disease relapses. Ninety-eight IBD patients in a state of clinical remission (66 UC, 32 CD) and 97 controls were recruited, and stool samples, as well as detailed patient data, were collected. After DNA extraction, the variable regions V1 and V2 of the 16S rRNA gene were amplified and sequenced. Patients with IBD had a decrease in alpha diversity compared to that of healthy controls, and the beta diversity indices showed dissimilarity between the cohorts. Healthy controls were associated with the beneficial organisms unclassified Akkermansia species (Akkermansia uncl.), Oscillibacter uncl., and Coprococcus uncl., while flavonoid-degrading bacteria were associated with IBD. Network analysis identified highly central and influential disease markers and a strongly correlated network module of Enterobacteriaceae which was associated with IBD and could act as drivers for residual inflammatory processes sustaining and triggering IBD, even in a state of low disease activity. The microbiota in IBD patients is significantly different from that of healthy controls, even in a state of remission, which implicates the microbiota as an important driver of chronicity in IBD. IMPORTANCE Dysbiosis in inflammatory bowel disease (IBD) has been implicated as a causal or contributory factor to the pathogenesis of the disease. This study, done on patients in remission while accounting for various confounding factors, shows significant community differences and altered community dynamics, even after acute inflammation has subsided. A cluster of Enterobacteriaceae was linked with Crohn's disease, suggesting that this cluster, which contains members known to disrupt colonization resistance and form biofilms, persists during quiescence and can lead to chronic inflammation. Flavonoid-degrading bacteria were also associated with IBD, raising the possibility that modification of dietary flavonoids might induce and maintain remission in IBD.
Keywords: Enterobacteriaceae; dysbiosis; flavonoid-degrading bacteria; inflammatory bowel disease; microbiota; remission.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
