

A Bayesian model selection approach to mediation analysis
- PMID: 35533209
- PMCID: PMC9129027
- DOI: 10.1371/journal.pgen.1010184
A Bayesian model selection approach to mediation analysis
Abstract
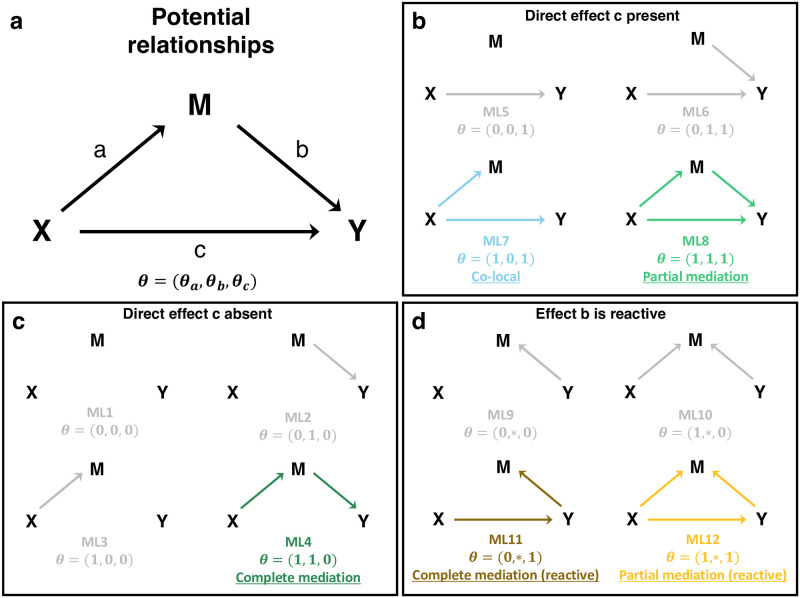
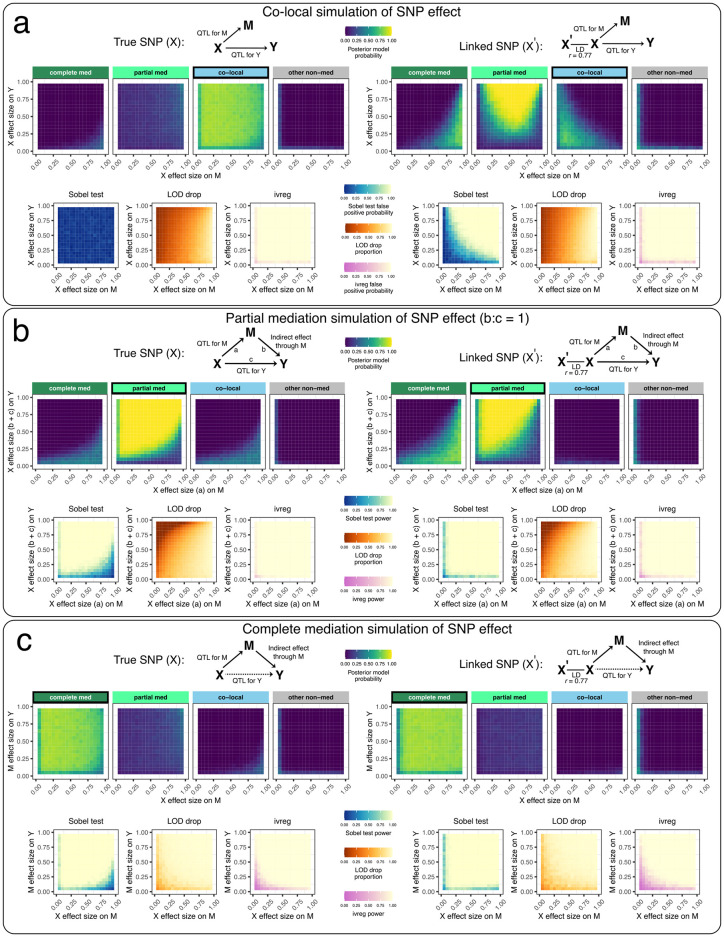
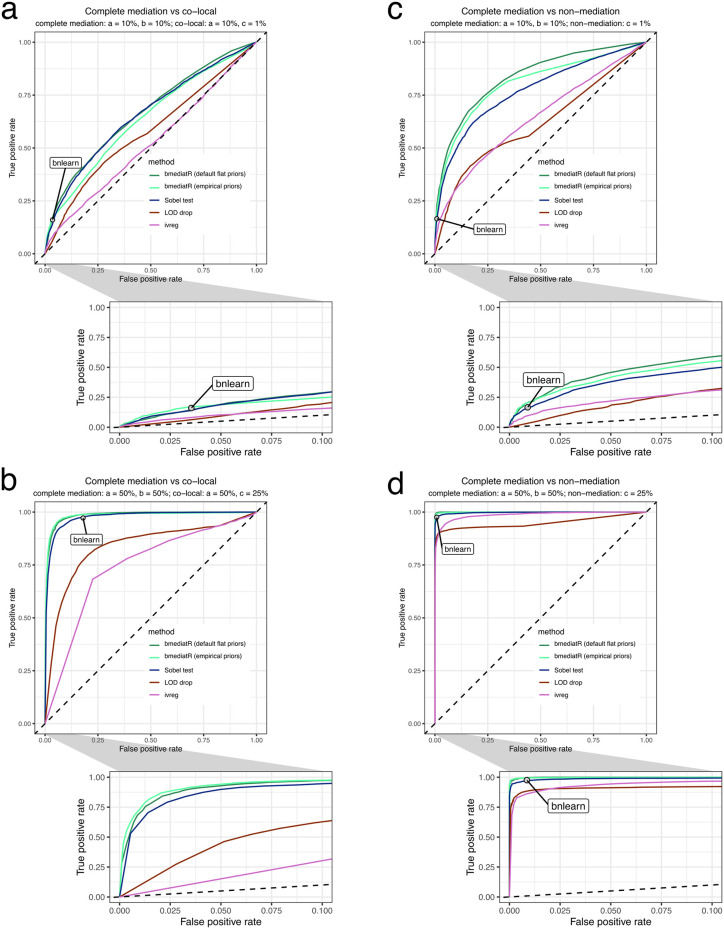
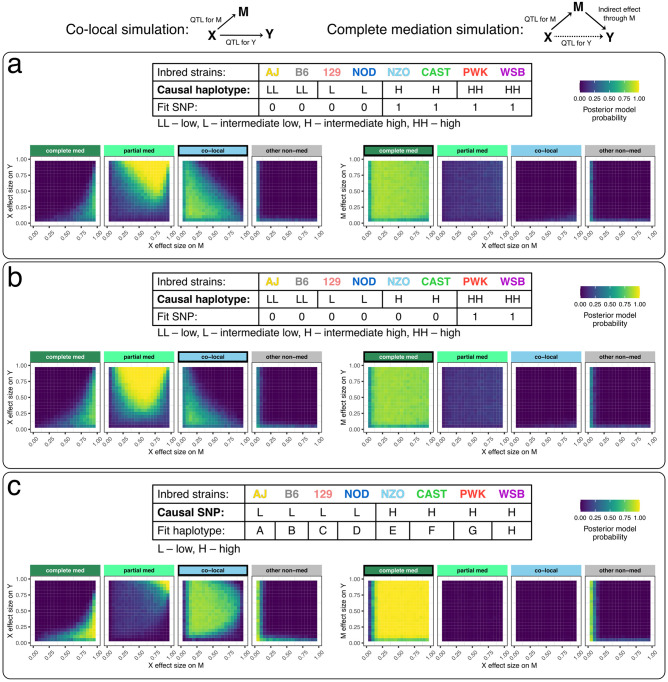
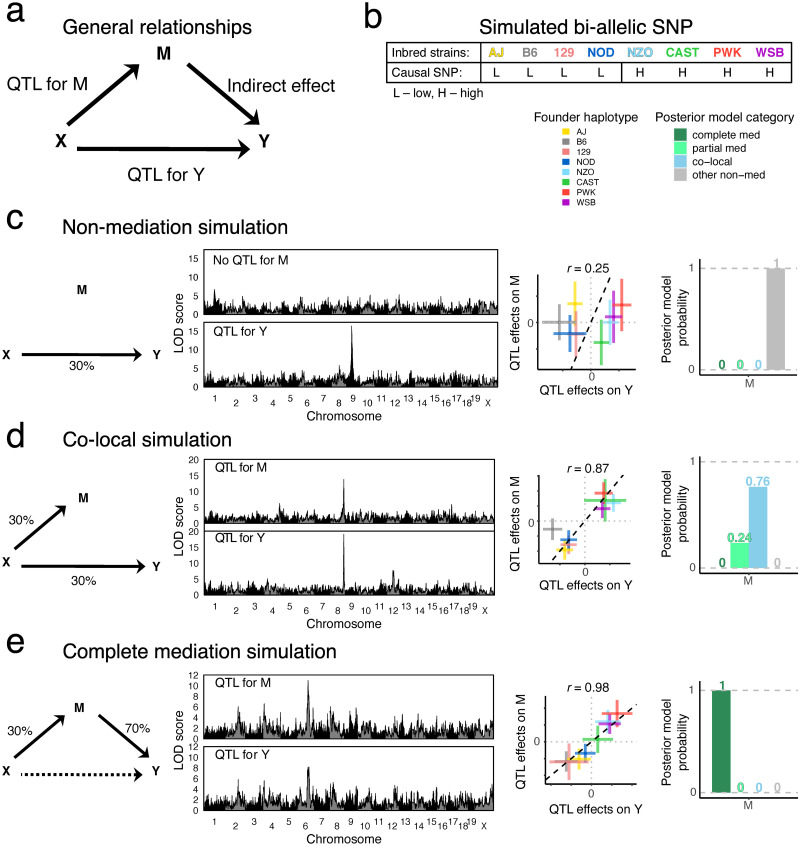
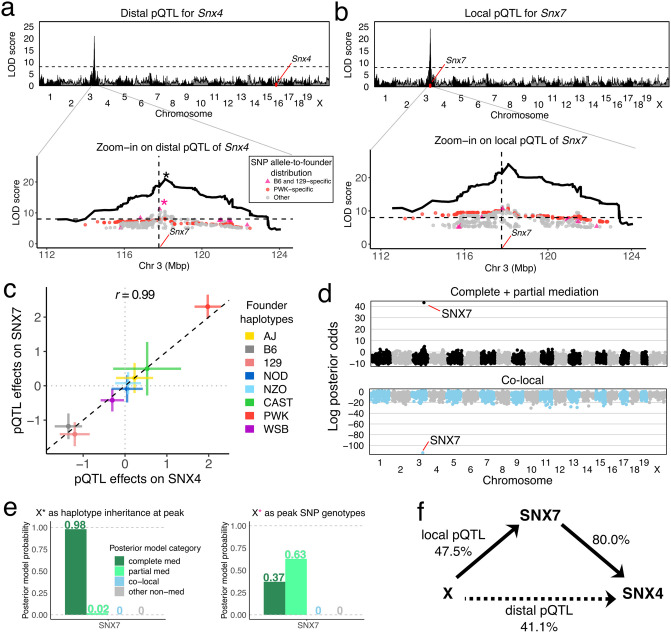
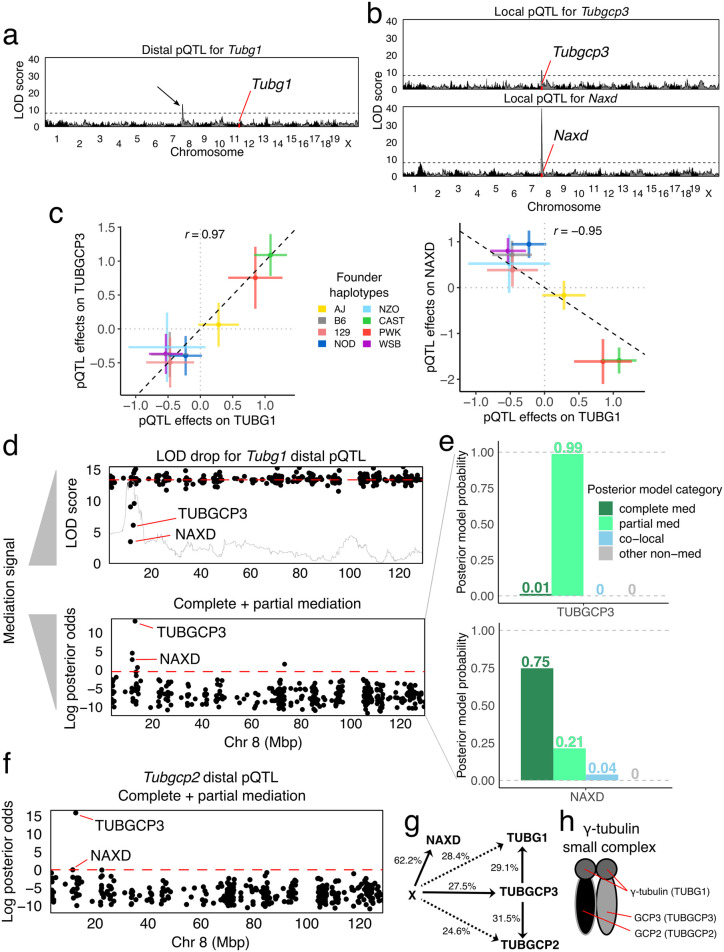
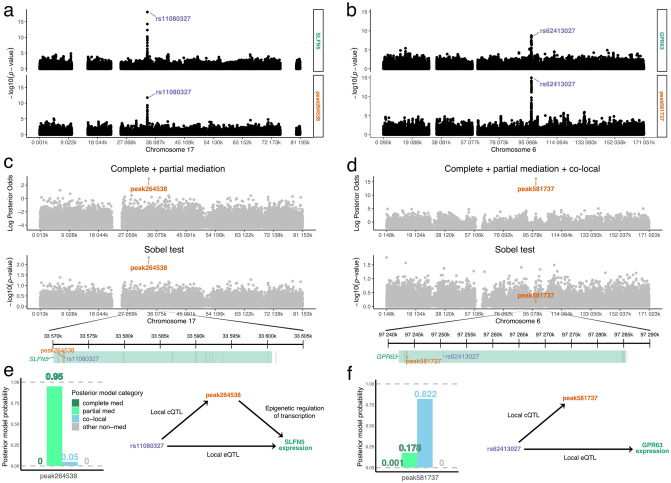
Genetic studies often seek to establish a causal chain of events originating from genetic variation through to molecular and clinical phenotypes. When multiple phenotypes share a common genetic association, one phenotype may act as an intermediate for the genetic effects on the other. Alternatively, the phenotypes may be causally unrelated but share genetic loci. Mediation analysis represents a class of causal inference approaches used to determine which of these scenarios is most plausible. We have developed a general approach to mediation analysis based on Bayesian model selection and have implemented it in an R package, bmediatR. Bayesian model selection provides a flexible framework that can be tailored to different analyses. Our approach can incorporate prior information about the likelihood of models and the strength of causal effects. It can also accommodate multiple genetic variants or multi-state haplotypes. Our approach reports posterior probabilities that can be useful in interpreting uncertainty among competing models. We compared bmediatR with other popular methods, including the Sobel test, Mendelian randomization, and Bayesian network analysis using simulated data. We found that bmediatR performed as well or better than these alternatives in most scenarios. We applied bmediatR to proteome data from Diversity Outbred (DO) mice, a multi-parent population, and demonstrate the power of mediation with multi-state haplotypes. We also applied bmediatR to data from human cell lines to identify transcripts that are mediated through or are expressed independently from local chromatin accessibility. We demonstrate that Bayesian model selection provides a powerful and versatile approach to identify causal relationships in genetic studies using model organism or human data.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Bayesian network-based Mendelian randomization for variant prioritization and phenotypic causal inference.Hum Genet. 2024 Oct;143(9-10):1081-1094. doi: 10.1007/s00439-024-02640-x. Epub 2024 Feb 21. Hum Genet. 2024. PMID: 38381161
-
Inferring the direction of a causal link and estimating its effect via a Bayesian Mendelian randomization approach.Stat Methods Med Res. 2020 Apr;29(4):1081-1111. doi: 10.1177/0962280219851817. Epub 2019 May 30. Stat Methods Med Res. 2020. PMID: 31146640 Free PMC article.
-
Profile-likelihood Bayesian model averaging for two-sample summary data Mendelian randomization in the presence of horizontal pleiotropy.Stat Med. 2022 Mar 15;41(6):1100-1119. doi: 10.1002/sim.9320. Epub 2022 Jan 20. Stat Med. 2022. PMID: 35060160 Free PMC article.
-
Application of Bayesian genomic prediction methods to genome-wide association analyses.Genet Sel Evol. 2022 May 13;54(1):31. doi: 10.1186/s12711-022-00724-8. Genet Sel Evol. 2022. PMID: 35562659 Free PMC article. Review.
-
Causal graphs for the analysis of genetic cohort data.Physiol Genomics. 2020 Sep 1;52(9):369-378. doi: 10.1152/physiolgenomics.00115.2019. Epub 2020 Jul 20. Physiol Genomics. 2020. PMID: 32687429 Free PMC article. Review.
Cited by
-
Transcripts with high distal heritability mediate genetic effects on complex metabolic traits.Nat Commun. 2025 Jul 1;16(1):5507. doi: 10.1038/s41467-025-61228-9. Nat Commun. 2025. PMID: 40595617 Free PMC article.
-
Transcripts with high distal heritability mediate genetic effects on complex metabolic traits.bioRxiv [Preprint]. 2024 Sep 27:2024.09.26.613931. doi: 10.1101/2024.09.26.613931. bioRxiv. 2024. Update in: Nat Commun. 2025 Jul 1;16(1):5507. doi: 10.1038/s41467-025-61228-9. PMID: 39386475 Free PMC article. Updated. Preprint.
-
Systems genetics approaches for understanding complex traits with relevance for human disease.Elife. 2023 Nov 14;12:e91004. doi: 10.7554/eLife.91004. Elife. 2023. PMID: 37962168 Free PMC article. Review.
-
Enhancer RNA Transcriptome-Wide Association Study Reveals a Distinctive Class of Pan-Cancer Susceptibility eRNAs.Adv Sci (Weinh). 2025 Apr;12(13):e2411974. doi: 10.1002/advs.202411974. Epub 2025 Feb 14. Adv Sci (Weinh). 2025. PMID: 39950845 Free PMC article.
-
Genetics of cell-type-specific post-transcriptional gene regulation during human neurogenesis.bioRxiv [Preprint]. 2023 Sep 1:2023.08.30.555019. doi: 10.1101/2023.08.30.555019. bioRxiv. 2023. Update in: Am J Hum Genet. 2024 Sep 5;111(9):1877-1898. doi: 10.1016/j.ajhg.2024.07.015. PMID: 37693528 Free PMC article. Updated. Preprint.
References
-
- Judd CM, Kenny DA. Data Analysis in Social Psychology: Recent and Recurring Issues. In: Fiske ST, Gilbert DT, Lindzey G, editors. Handbook of Social Psychology. Hoboken, N.J.: American Cancer Society; 2010. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/9780470561119.socpsy001004. - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
