

Transcriptional Integration of Distinct Microbial and Nutritional Signals by the Small Intestinal Epithelium
- PMID: 35533983
- PMCID: PMC9305020
- DOI: 10.1016/j.jcmgh.2022.04.013
Transcriptional Integration of Distinct Microbial and Nutritional Signals by the Small Intestinal Epithelium
Abstract
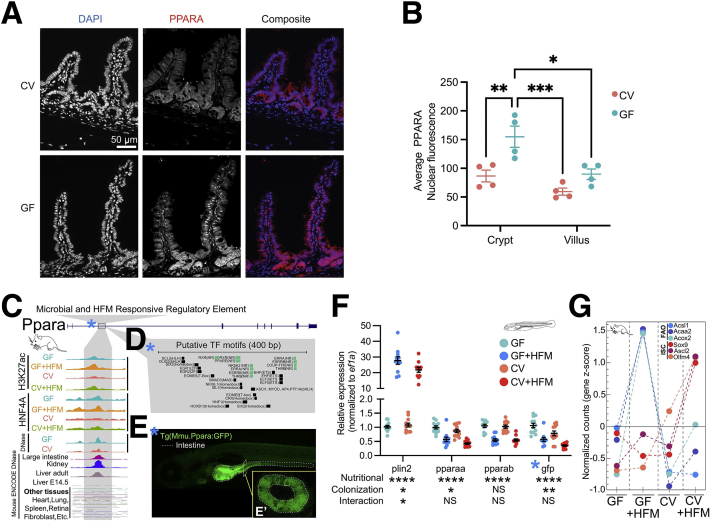
Background & aims: The intestine constantly interprets and adapts to complex combinations of dietary and microbial stimuli. However, the transcriptional strategies by which the intestinal epithelium integrates these coincident sources of information remain unresolved. We recently found that microbiota colonization suppresses epithelial activity of hepatocyte nuclear factor 4 nuclear receptor transcription factors, but their integrative regulation was unknown.
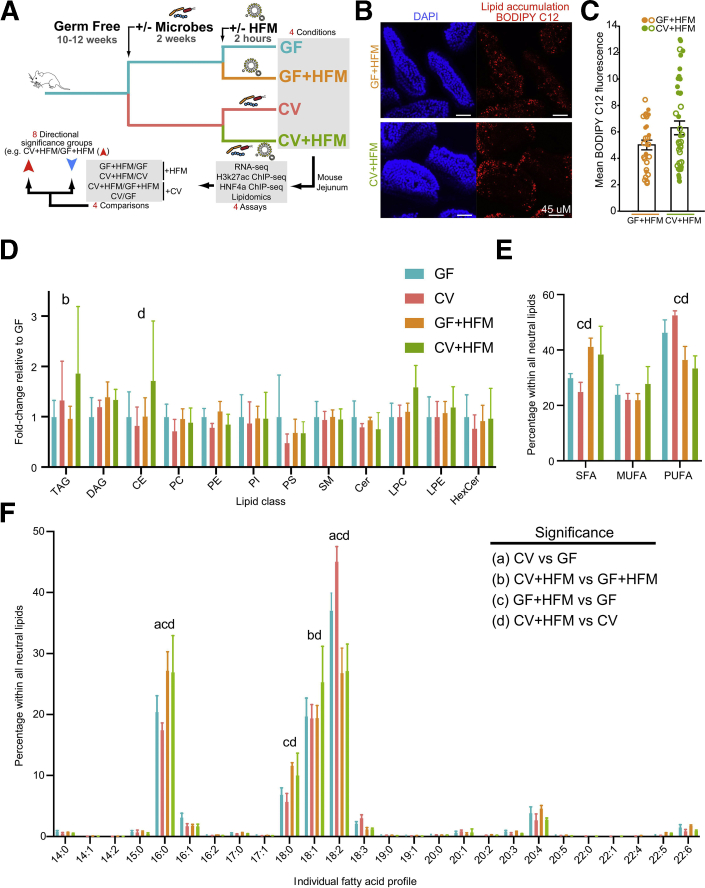
Methods: We compared adult mice reared germ-free or conventionalized with a microbiota either fed normally or after a single high-fat meal. Preparations of unsorted jejunal intestinal epithelial cells were queried using lipidomics and genome-wide assays for RNA sequencing and ChIP sequencing for the activating histone mark H3K27ac and hepatocyte nuclear factor 4 alpha.
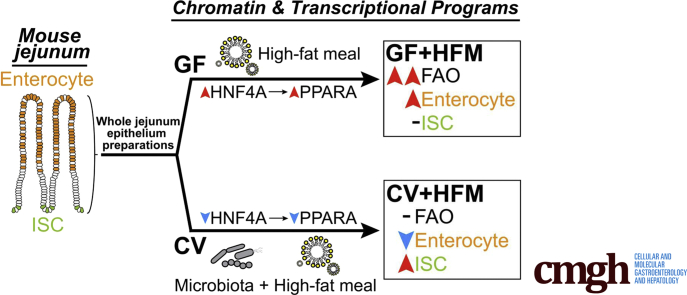
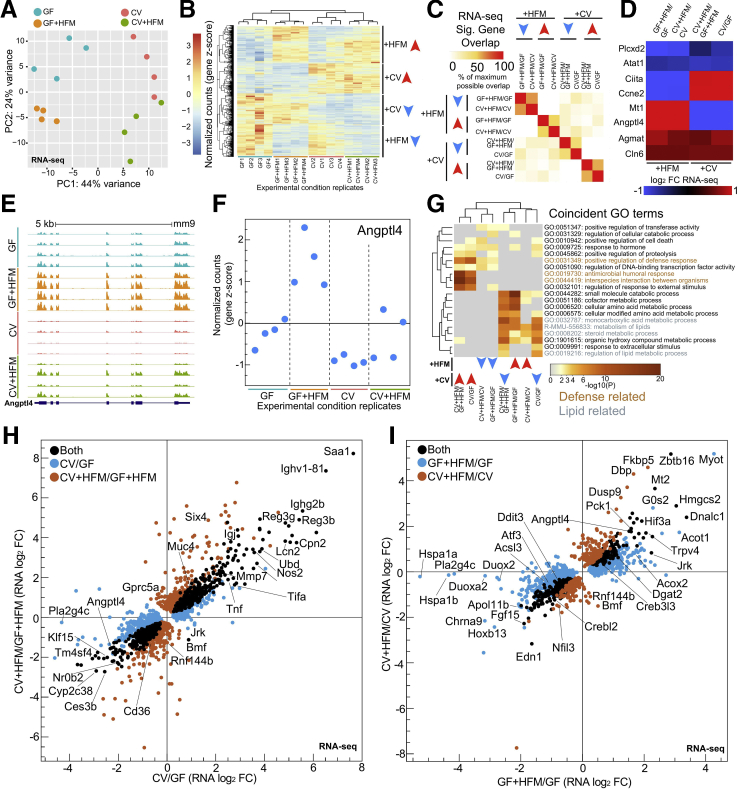
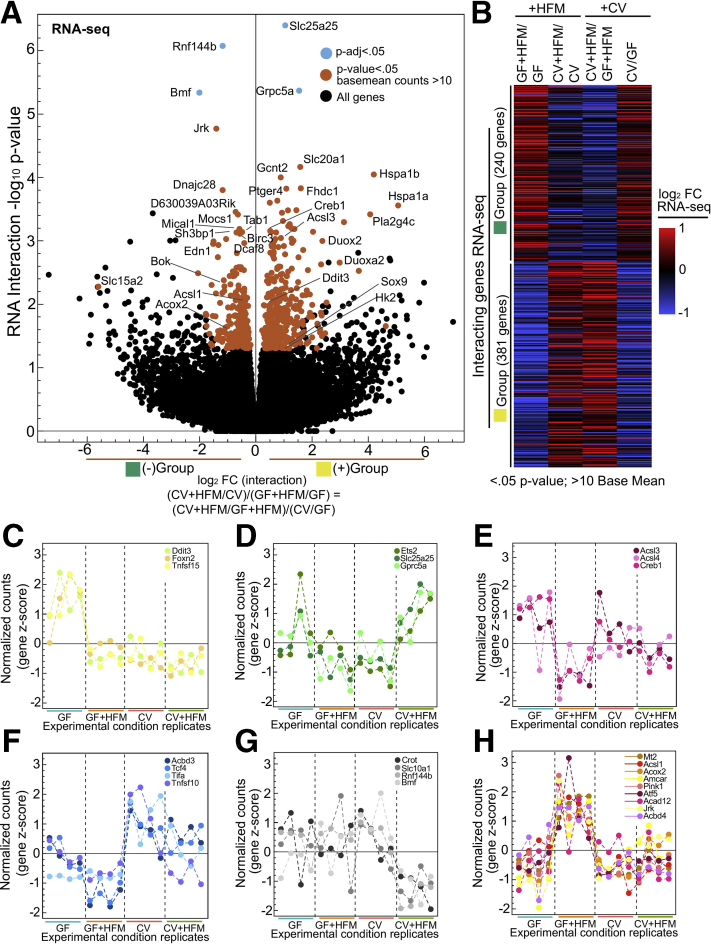
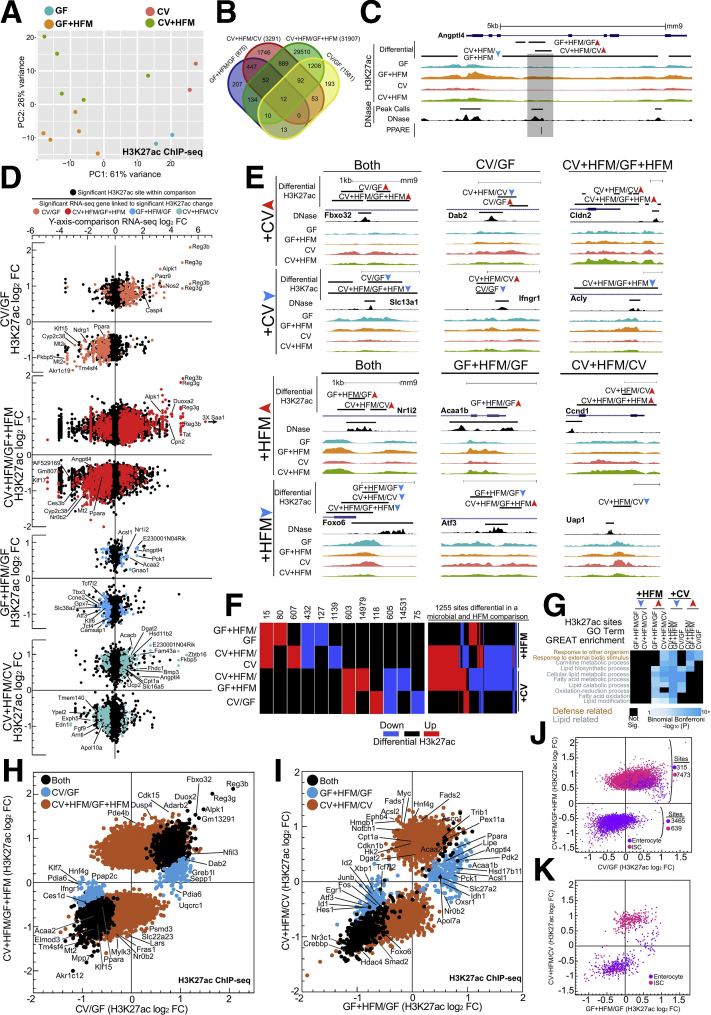
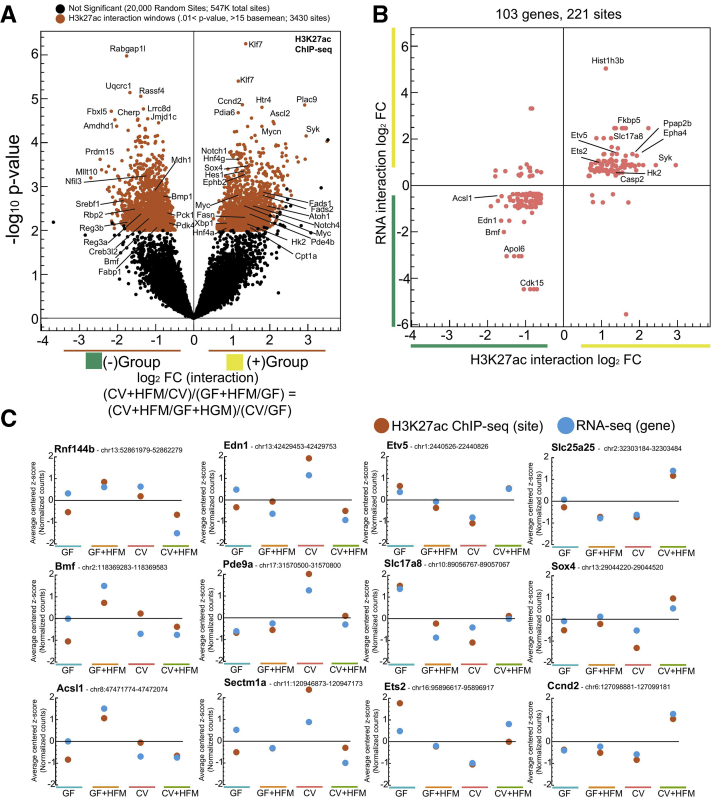
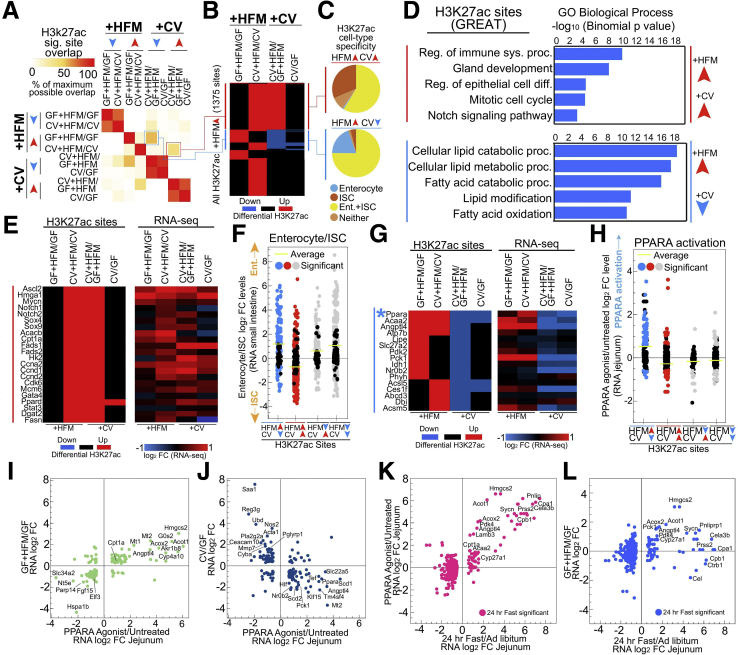
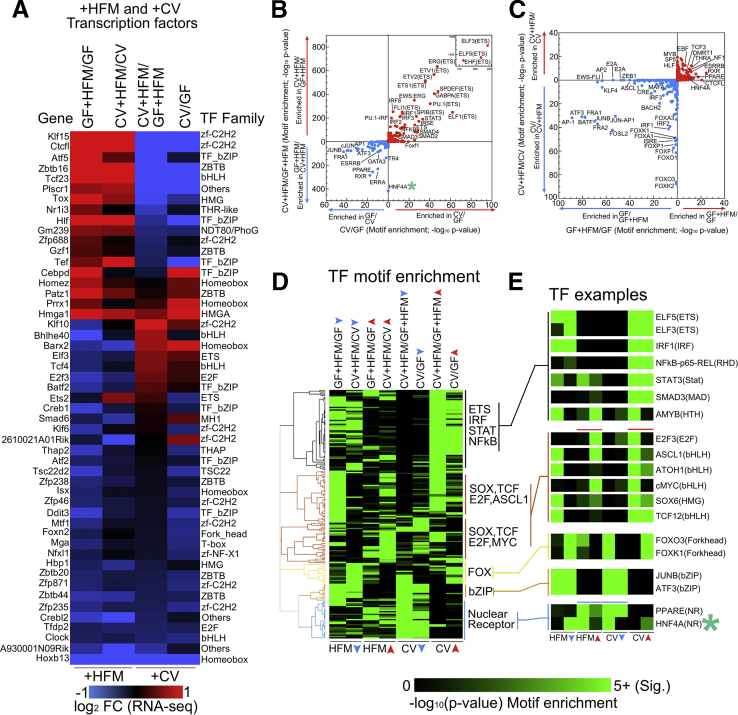
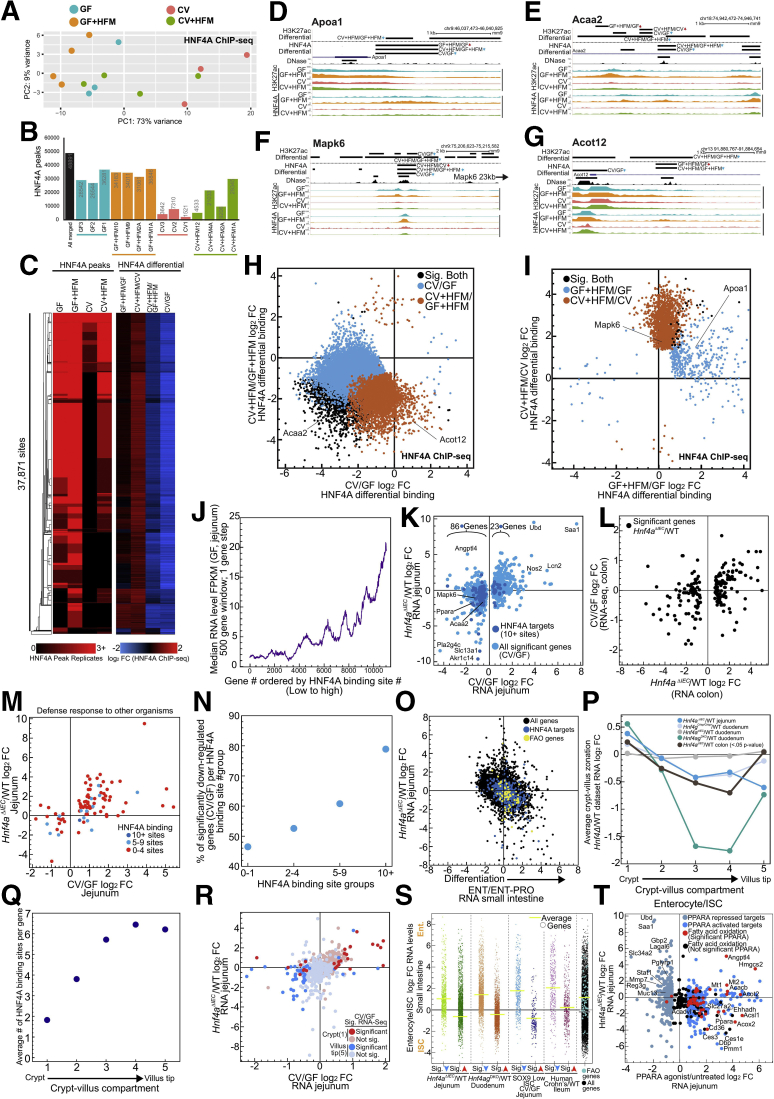
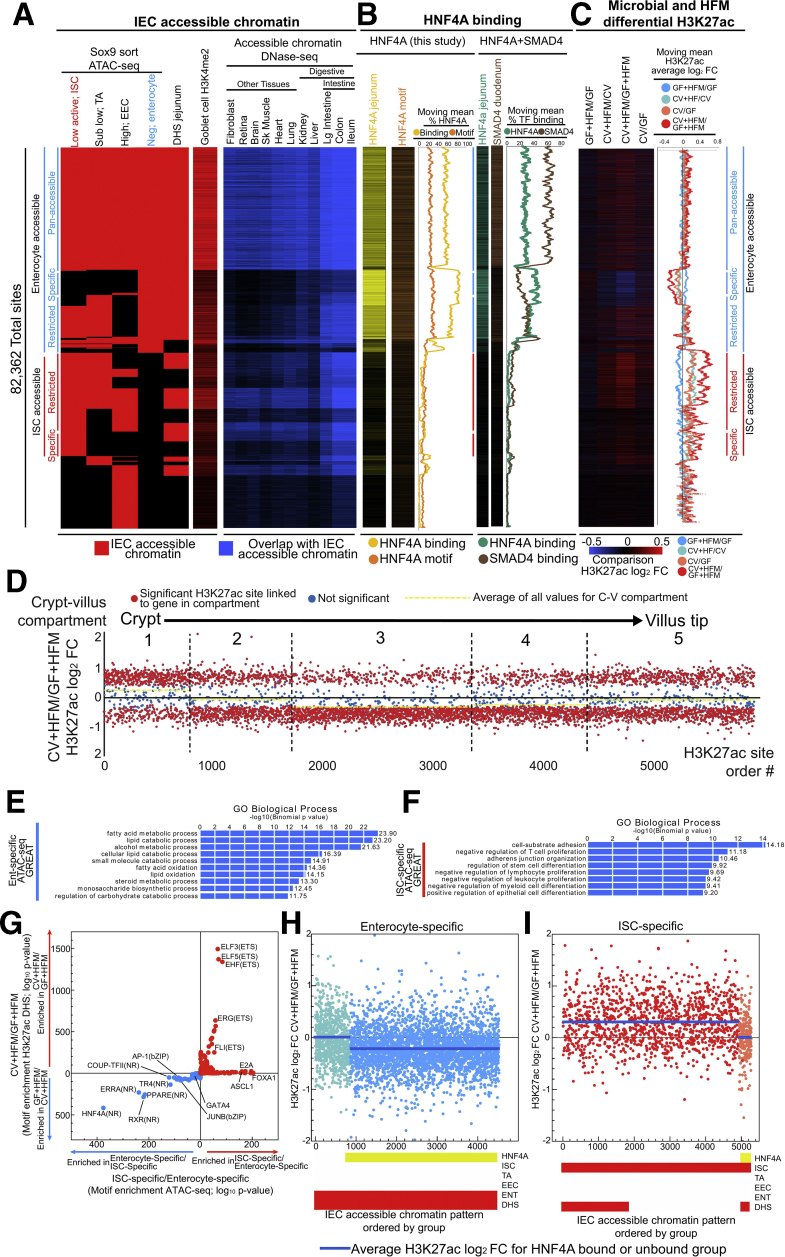
Results: Analysis of lipid classes, genes, and regulatory regions identified distinct nutritional and microbial responses but also simultaneous influence of both stimuli. H3K27ac sites preferentially increased by high-fat meal in the presence of microbes neighbor lipid anabolism and proliferation genes, were previously identified intestinal stem cell regulatory regions, and were not hepatocyte nuclear factor 4 alpha targets. In contrast, H3K27ac sites preferentially increased by high-fat meal in the absence of microbes neighbor targets of the energy homeostasis regulator peroxisome proliferator activated receptor alpha, neighbored fatty acid oxidation genes, were previously identified enterocyte regulatory regions, and were hepatocyte factor 4 alpha bound.
Conclusions: Hepatocyte factor 4 alpha supports a differentiated enterocyte and fatty acid oxidation program in germ-free mice, and that suppression of hepatocyte factor 4 alpha by the combination of microbes and high-fat meal may result in preferential activation of intestinal epithelial cell proliferation programs. This identifies potential transcriptional mechanisms for intestinal adaptation to multiple signals and how microbiota may modulate intestinal lipid absorption, epithelial cell renewal, and systemic energy balance.
Keywords: Chromatin; Intestine; Lipid Metabolism; Microbiome.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Martinez-Guryn K., Hubert N., Frazier K., Urlass S., Musch M.W., Ojeda P., Pierre J.F., Miyoshi J., Sontag T.J., Cham C.M., Reardon C.A., Leone V., Chang E.B. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe. 2018;23:458–469 e5. - PMC - PubMed
-
- Patnode M.L., Beller Z.W., Han N.D., Cheng J., Peters S.L., Terrapon N., Henrissat B., Le Gall S., Saulnier L., Hayashi D.K., Meynier A., Vinoy S., Giannone R.J., Hettich R.L., Gordon J.I. Interspecies competition impacts targeted manipulation of human gut bacteria by fiber-derived glycans. Cell. 2019;179:59–73 e13. - PMC - PubMed
-
- Krautkramer K.A., Fan J., Backhed F. Gut microbial metabolites as multi-kingdom intermediates. Nat Rev Microbiol. 2021;19:77–94. - PubMed
-
- Byndloss M.X., Olsan E.E., Rivera-Chavez F., Tiffany C.R., Cevallos S.A., Lokken K.L., Torres T.P., Byndloss A.J., Faber F., Gao Y., Litvak Y., Lopez C.A., Xu G., Napoli E., Giulivi C., Tsolis R.M., Revzin A., Lebrilla C.B., Baumler A.J. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. 2017;357:570–575. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
