

Spatial epitranscriptomics reveals A-to-I editome specific to cancer stem cell microniches
- PMID: 35534484
- PMCID: PMC9085828
- DOI: 10.1038/s41467-022-30299-3
Spatial epitranscriptomics reveals A-to-I editome specific to cancer stem cell microniches
Abstract
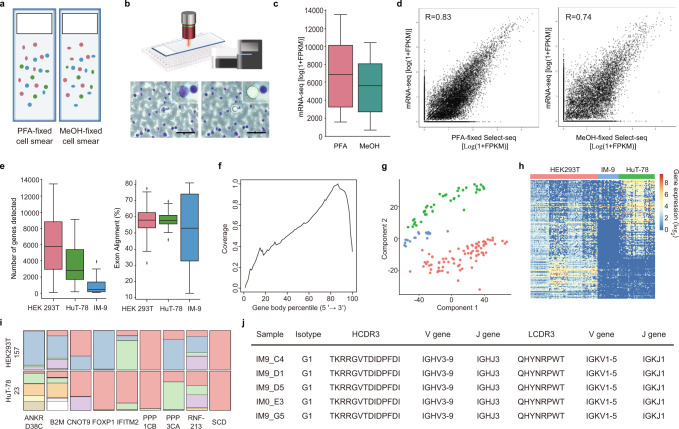
Epitranscriptomic features, such as single-base RNA editing, are sources of transcript diversity in cancer, but little is understood in terms of their spatial context in the tumour microenvironment. Here, we introduce spatial-histopathological examination-linked epitranscriptomics converged to transcriptomics with sequencing (Select-seq), which isolates regions of interest from immunofluorescence-stained tissue and obtains transcriptomic and epitranscriptomic data. With Select-seq, we analyse the cancer stem cell-like microniches in relation to the tumour microenvironment of triple-negative breast cancer patients. We identify alternative splice variants, perform complementarity-determining region analysis of infiltrating T cells and B cells, and assess adenosine-to-inosine base editing in tumour tissue sections. Especially, in triple-negative breast cancer microniches, adenosine-to-inosine editome specific to different microniche groups is identified.
© 2022. The Author(s).
Conflict of interest statement
A.C.L., Y.L., O.K., and S.K. are listed as inventors on patents related to the work applied by the Seoul National University covering the technology (Methods for selectively separating samples from substrate, US 15/770, 765). H.B.L. and W.H. report being a member on the board of directors of and holding stock and ownership interests at DCGen, Co., Ltd., not relevant to this study. The remaining authors declare no competing interests.
Figures





Comment in
-
Epitranscriptomics of cancer microniches.Nat Rev Cancer. 2023 Apr;23(4):189. doi: 10.1038/s41568-023-00552-y. Nat Rev Cancer. 2023. PMID: 36797378 No abstract available.
Similar articles
-
Suppression of adenosine-to-inosine (A-to-I) RNA editome by death associated protein 3 (DAP3) promotes cancer progression.Sci Adv. 2020 Jun 17;6(25):eaba5136. doi: 10.1126/sciadv.aba5136. eCollection 2020 Jun. Sci Adv. 2020. PMID: 32596459 Free PMC article.
-
The Cancer Editome Atlas: A Resource for Exploratory Analysis of the Adenosine-to-Inosine RNA Editome in Cancer.Cancer Res. 2019 Jun 1;79(11):3001-3006. doi: 10.1158/0008-5472.CAN-18-3501. Epub 2019 Apr 23. Cancer Res. 2019. PMID: 31015229
-
Adenosine-to-Inosine RNA Editing Enzyme ADAR and microRNAs.Methods Mol Biol. 2021;2181:83-95. doi: 10.1007/978-1-0716-0787-9_6. Methods Mol Biol. 2021. PMID: 32729076
-
Adenosine-to-inosine RNA editing meets cancer.Carcinogenesis. 2011 Nov;32(11):1569-77. doi: 10.1093/carcin/bgr124. Epub 2011 Jun 29. Carcinogenesis. 2011. PMID: 21715563 Review.
-
Editing of messenger RNA precursors and of tRNAs by adenosine to inosine conversion.FEBS Lett. 1999 Jun 4;452(1-2):71-6. doi: 10.1016/s0014-5793(99)00590-6. FEBS Lett. 1999. PMID: 10376681 Review.
Cited by
-
Epitranscriptomics in the development, functions, and disorders of cancer stem cells.Front Oncol. 2023 Mar 17;13:1145766. doi: 10.3389/fonc.2023.1145766. eCollection 2023. Front Oncol. 2023. PMID: 37007137 Free PMC article. Review.
-
Epitranscriptomics of cancer microniches.Nat Rev Cancer. 2023 Apr;23(4):189. doi: 10.1038/s41568-023-00552-y. Nat Rev Cancer. 2023. PMID: 36797378 No abstract available.
-
Barcoded multiple displacement amplification for high coverage sequencing in spatial genomics.Nat Commun. 2023 Aug 29;14(1):5261. doi: 10.1038/s41467-023-41019-w. Nat Commun. 2023. PMID: 37644058 Free PMC article.
-
Fatty Acid Metabolites and the Tumor Microenvironment as Potent Regulators of Cancer Stem Cell Signaling.Metabolites. 2023 May 31;13(6):709. doi: 10.3390/metabo13060709. Metabolites. 2023. PMID: 37367867 Free PMC article. Review.
-
Mapping cancer biology in space: applications and perspectives on spatial omics for oncology.Mol Cancer. 2024 Jan 30;23(1):26. doi: 10.1186/s12943-024-01941-z. Mol Cancer. 2024. PMID: 38291400 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
