

Massively parallel identification of functionally consequential noncoding genetic variants in undiagnosed rare disease patients
- PMID: 35534523
- PMCID: PMC9085742
- DOI: 10.1038/s41598-022-11589-8
Massively parallel identification of functionally consequential noncoding genetic variants in undiagnosed rare disease patients
Abstract
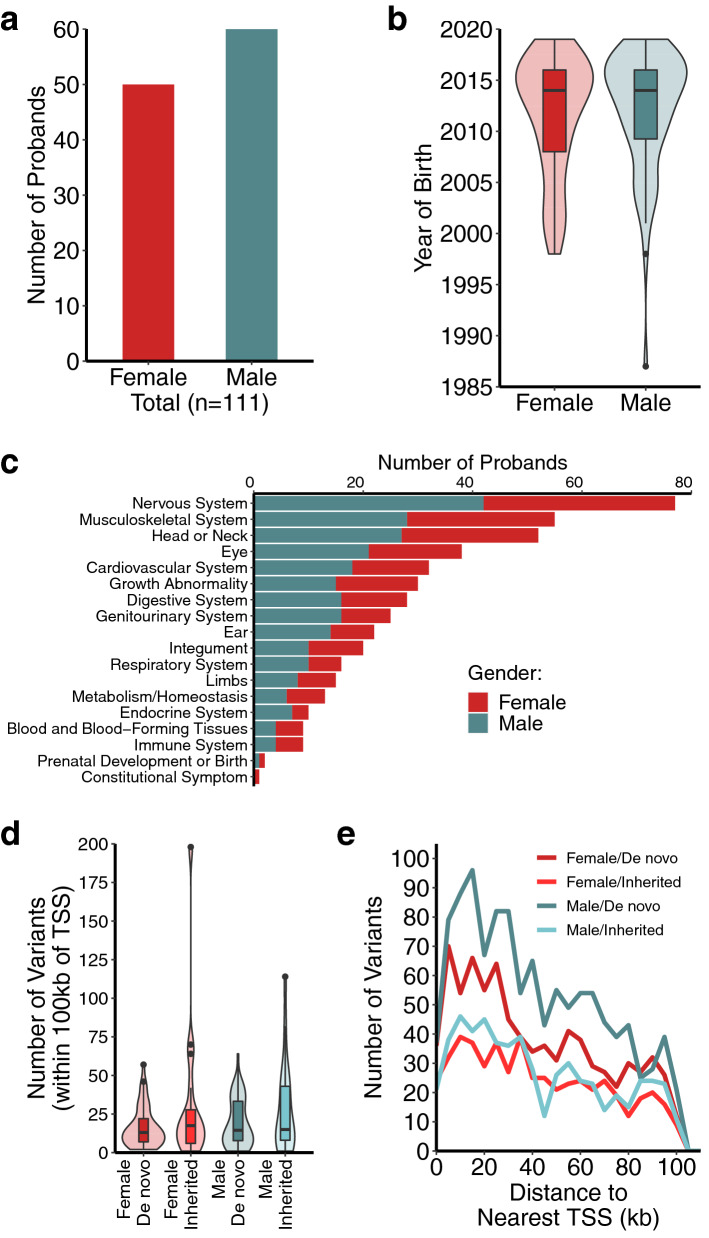
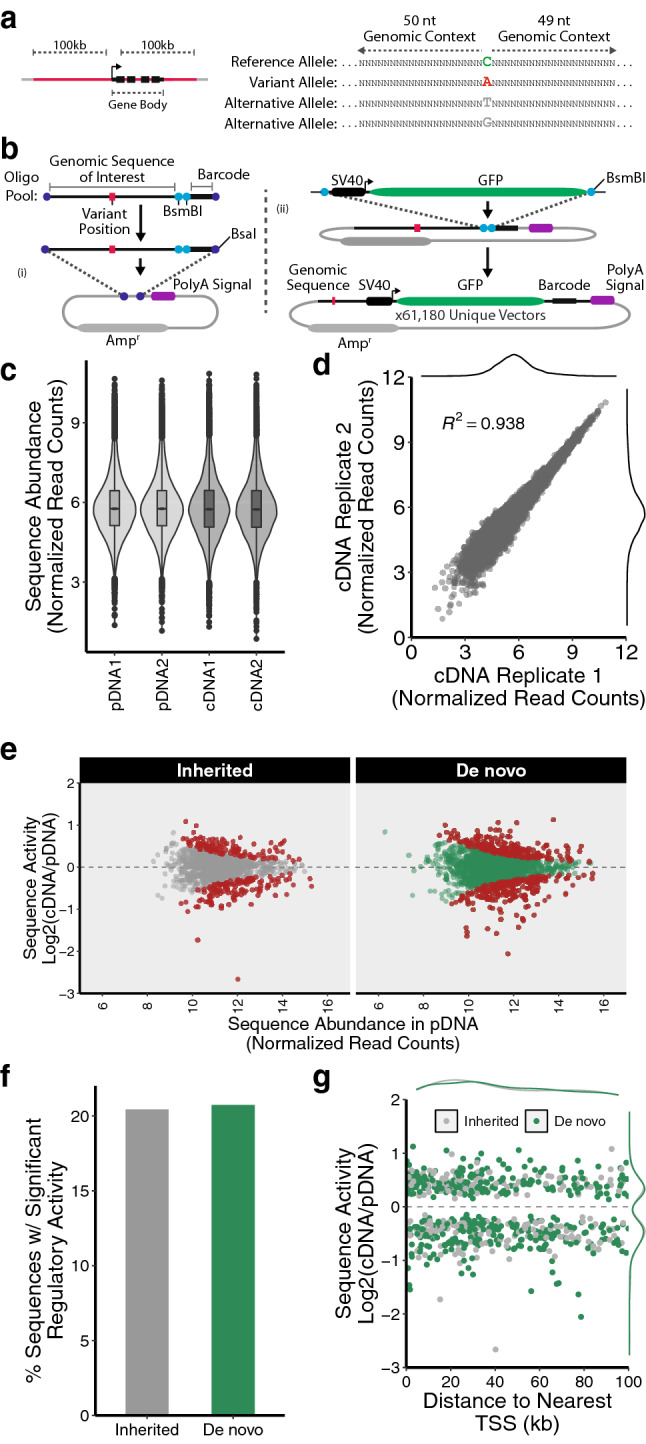
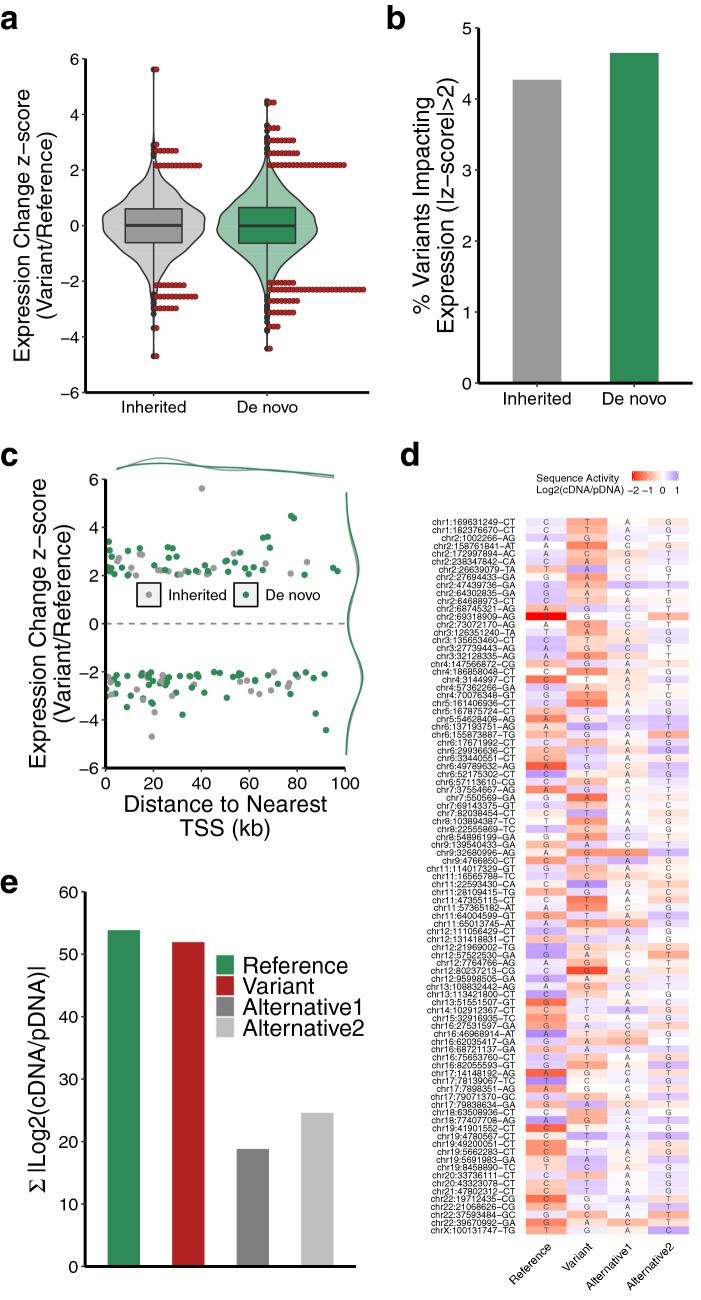
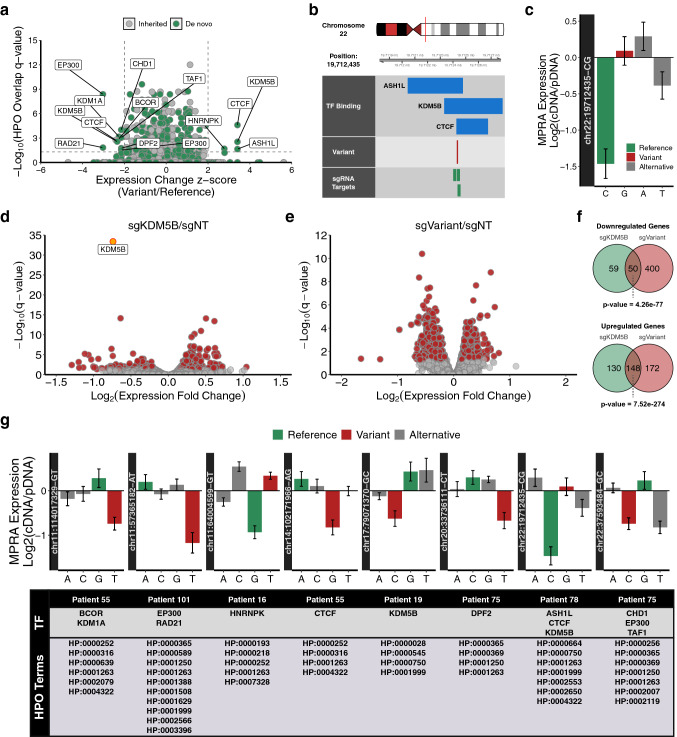
Clinical whole genome sequencing has enabled the discovery of potentially pathogenic noncoding variants in the genomes of rare disease patients with a prior history of negative genetic testing. However, interpreting the functional consequences of noncoding variants and distinguishing those that contribute to disease etiology remains a challenge. Here we address this challenge by experimentally profiling the functional consequences of rare noncoding variants detected in a cohort of undiagnosed rare disease patients at scale using a massively parallel reporter assay. We demonstrate that this approach successfully identifies rare noncoding variants that alter the regulatory capacity of genomic sequences. In addition, we describe an integrative analysis that utilizes genomic features alongside patient clinical data to further prioritize candidate variants with an increased likelihood of pathogenicity. This work represents an important step towards establishing a framework for the functional interpretation of clinically detected noncoding variants.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
