

Within-sibship genome-wide association analyses decrease bias in estimates of direct genetic effects
- PMID: 35534559
- PMCID: PMC9110300
- DOI: 10.1038/s41588-022-01062-7
Within-sibship genome-wide association analyses decrease bias in estimates of direct genetic effects
Abstract
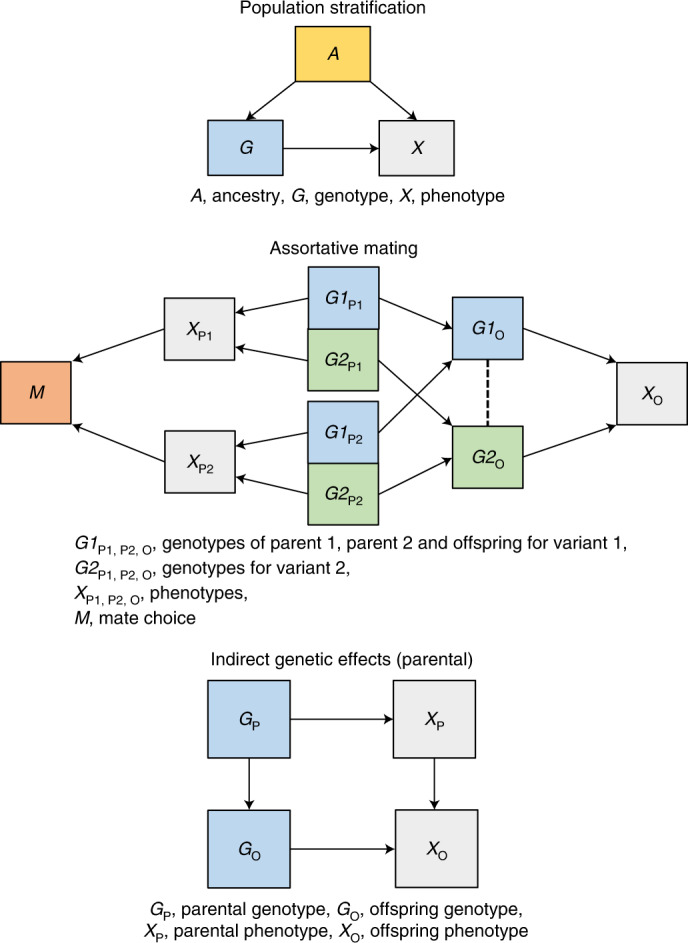
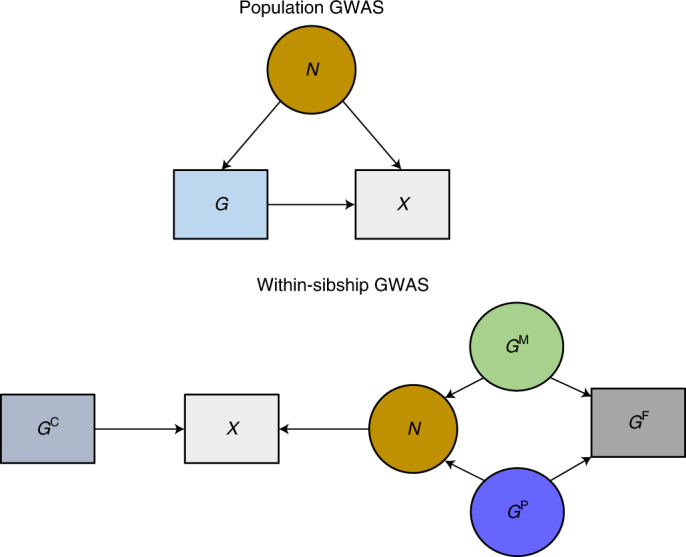
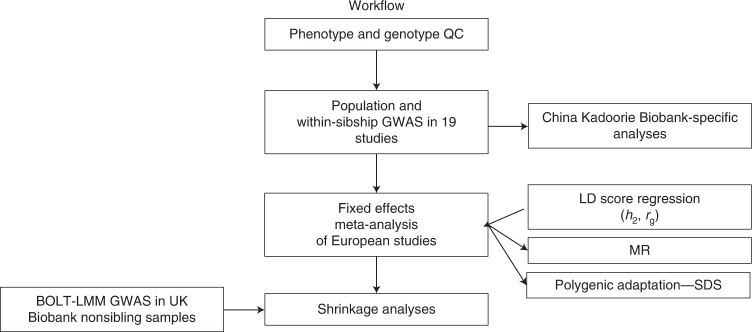
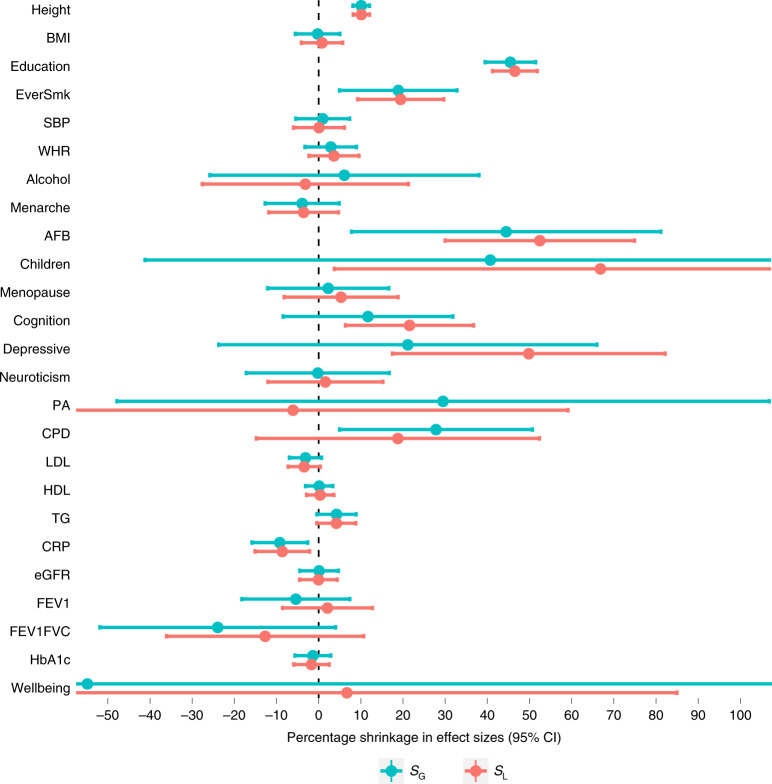
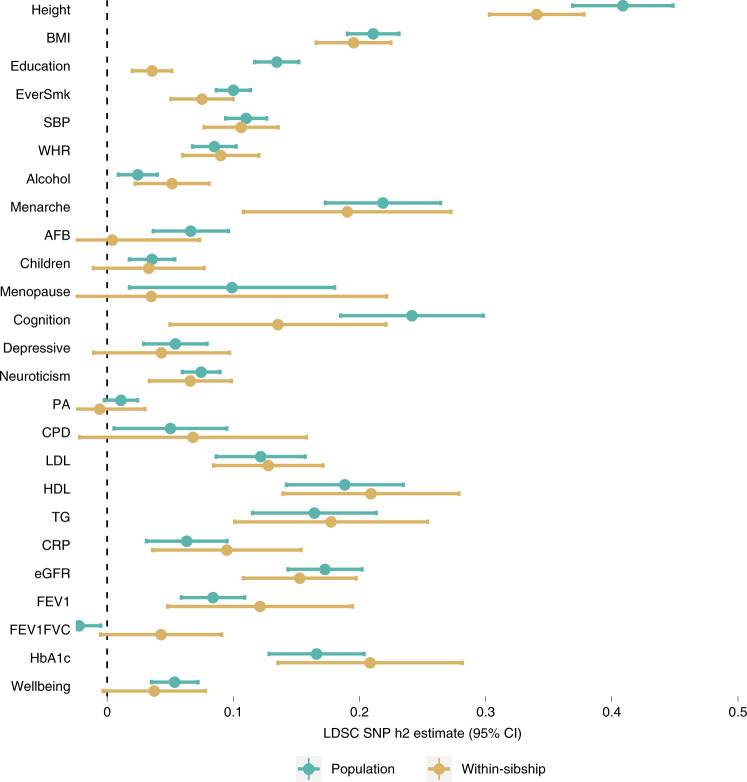
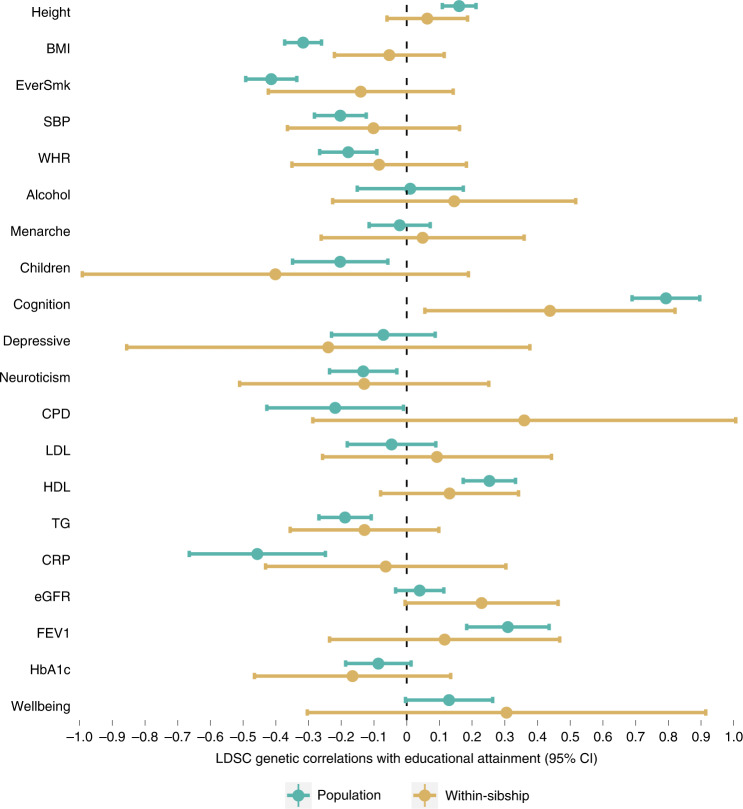
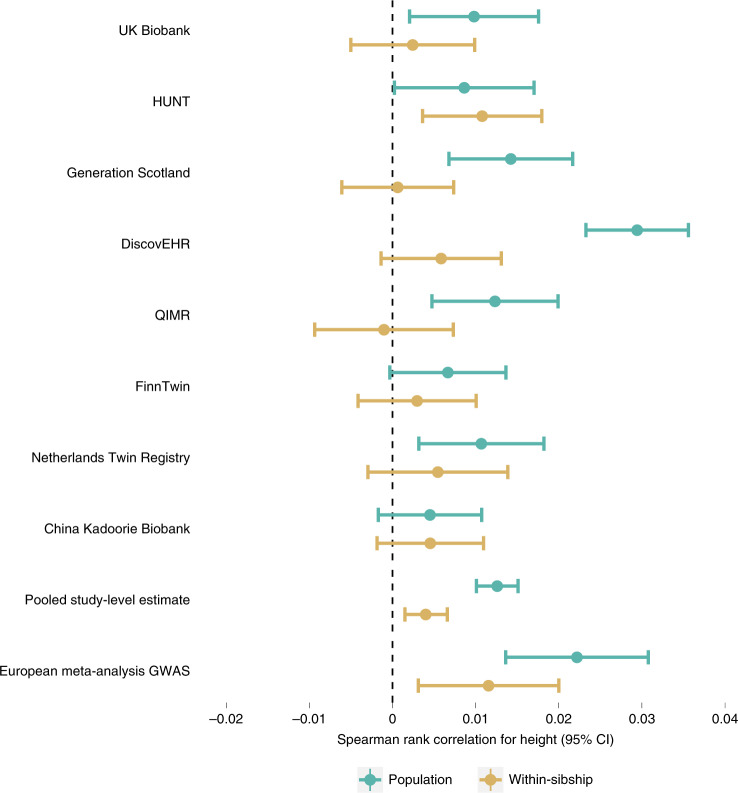
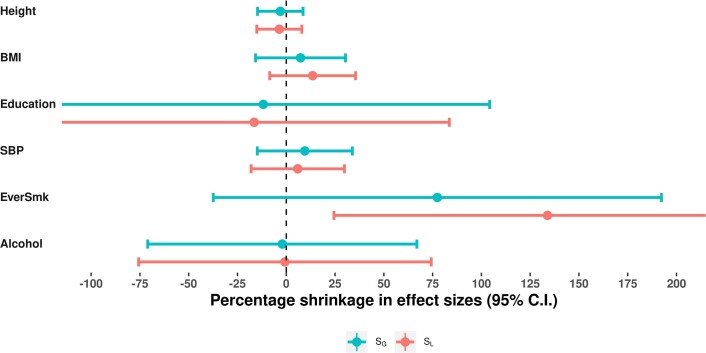
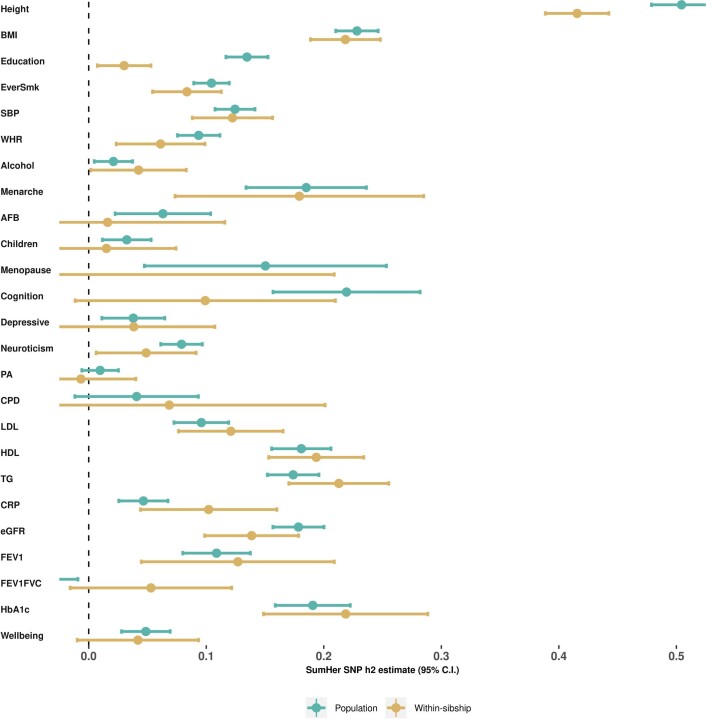
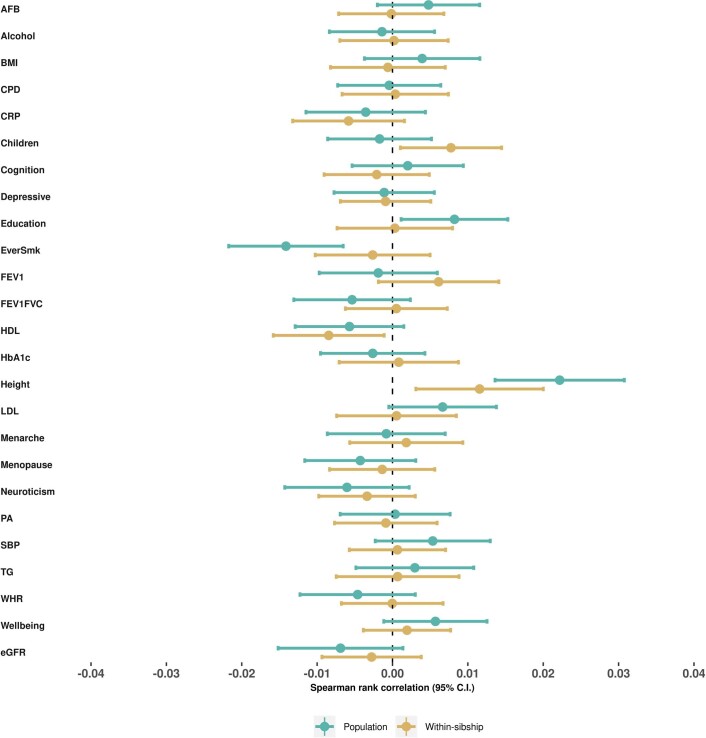
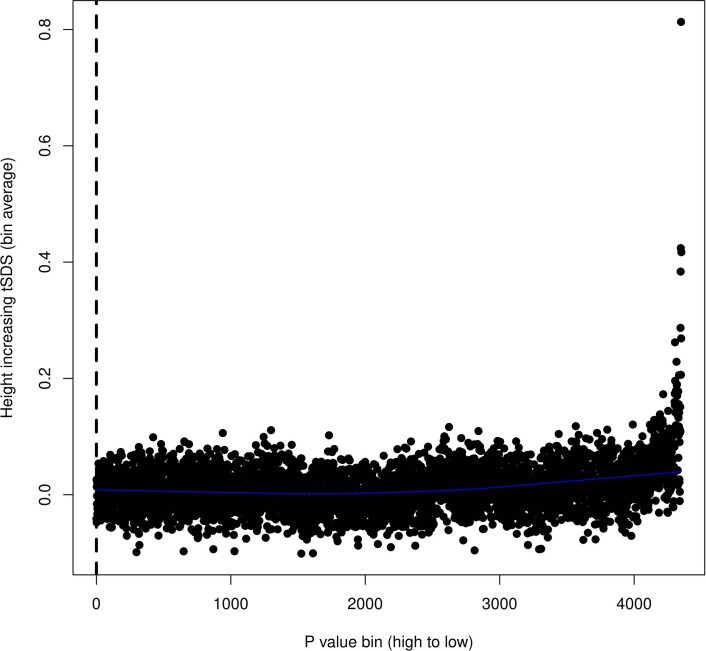
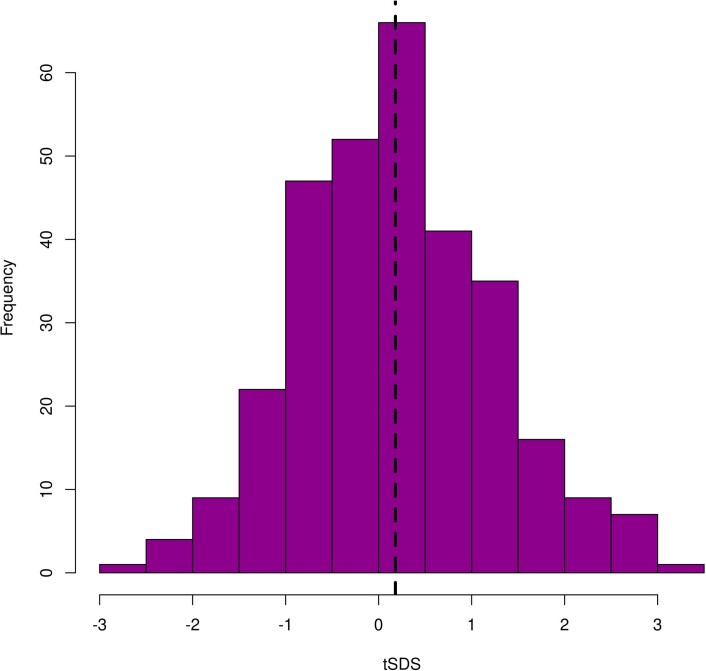
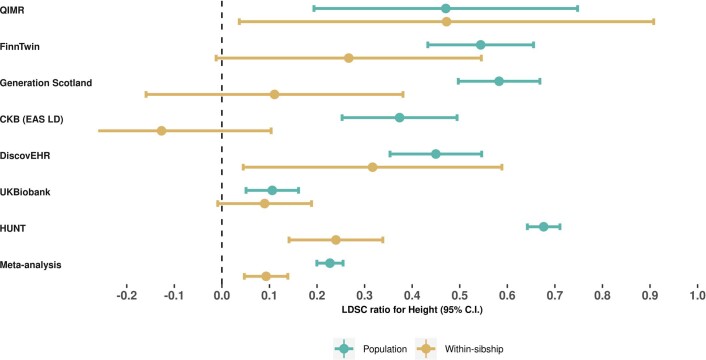
Estimates from genome-wide association studies (GWAS) of unrelated individuals capture effects of inherited variation (direct effects), demography (population stratification, assortative mating) and relatives (indirect genetic effects). Family-based GWAS designs can control for demographic and indirect genetic effects, but large-scale family datasets have been lacking. We combined data from 178,086 siblings from 19 cohorts to generate population (between-family) and within-sibship (within-family) GWAS estimates for 25 phenotypes. Within-sibship GWAS estimates were smaller than population estimates for height, educational attainment, age at first birth, number of children, cognitive ability, depressive symptoms and smoking. Some differences were observed in downstream SNP heritability, genetic correlations and Mendelian randomization analyses. For example, the within-sibship genetic correlation between educational attainment and body mass index attenuated towards zero. In contrast, analyses of most molecular phenotypes (for example, low-density lipoprotein-cholesterol) were generally consistent. We also found within-sibship evidence of polygenic adaptation on taller height. Here, we illustrate the importance of family-based GWAS data for phenotypes influenced by demographic and indirect genetic effects.
© 2022. The Author(s).
Conflict of interest statement
O.A.A. is a consultant to HealthLytix in a capacity unrelated to this work. All other authors declare no competing interests.
Figures



References
-
- Risch, N. & Merikangas, K. The future of genetic studies of complex human diseases. Science273, 1516–1517 (1996). - PubMed
-
- Fisher, R. A. The Genetical Theory of Natural Selection (Oxford Univ. Press, 1930).
Publication types
MeSH terms
Grants and funding
- MC_UU_00011/6/MRC_/Medical Research Council/United Kingdom
- R01 HL109946/HL/NHLBI NIH HHS/United States
- MR/R026408/1/MRC_/Medical Research Council/United Kingdom
- 208806/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- MC_UU_12026/2/MRC_/Medical Research Council/United Kingdom
- MC_PC_14135/MRC_/Medical Research Council/United Kingdom
- MC_UU_00017/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00011/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_13049/MRC_/Medical Research Council/United Kingdom
- MC_UU_00007/10/MRC_/Medical Research Council/United Kingdom
- MC_U137686851/MRC_/Medical Research Council/United Kingdom
- R24 AG065184/AG/NIA NIH HHS/United States
- 212946/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- P2C HD042849/HD/NICHD NIH HHS/United States
- R56 AG042568/AG/NIA NIH HHS/United States
- R01 AG042568/AG/NIA NIH HHS/United States
- MR/T030852/1/MRC_/Medical Research Council/United Kingdom
