

Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities
- PMID: 35534562
- PMCID: PMC9374001
- DOI: 10.1038/s41588-022-01061-8
Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities
Abstract
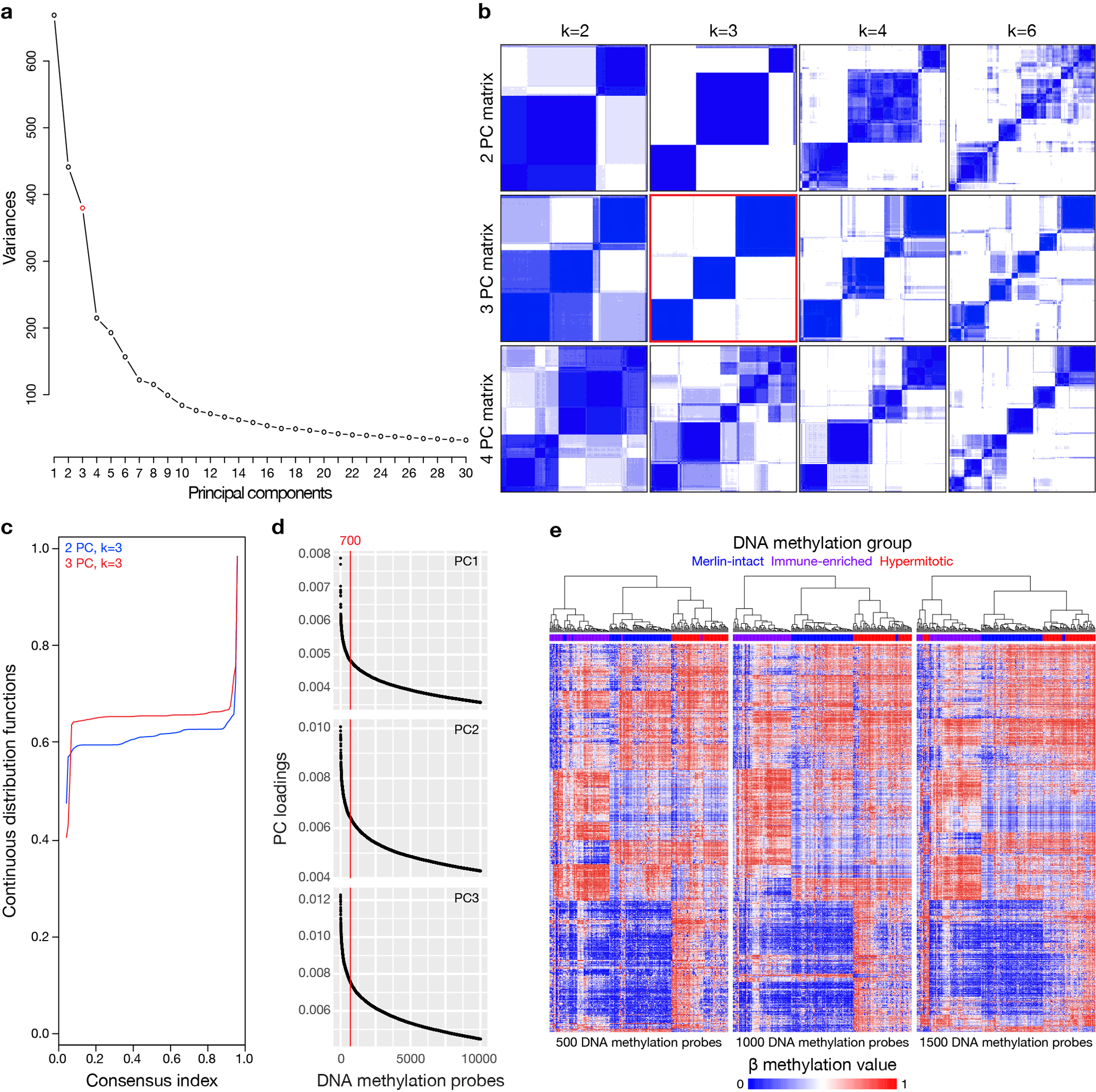
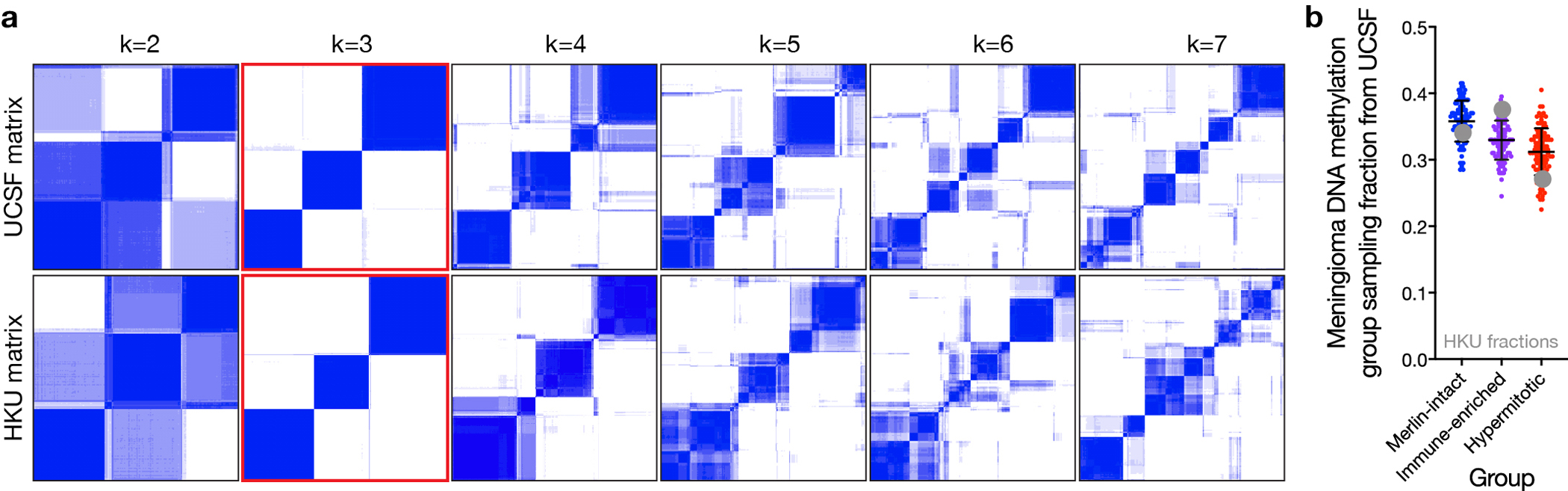
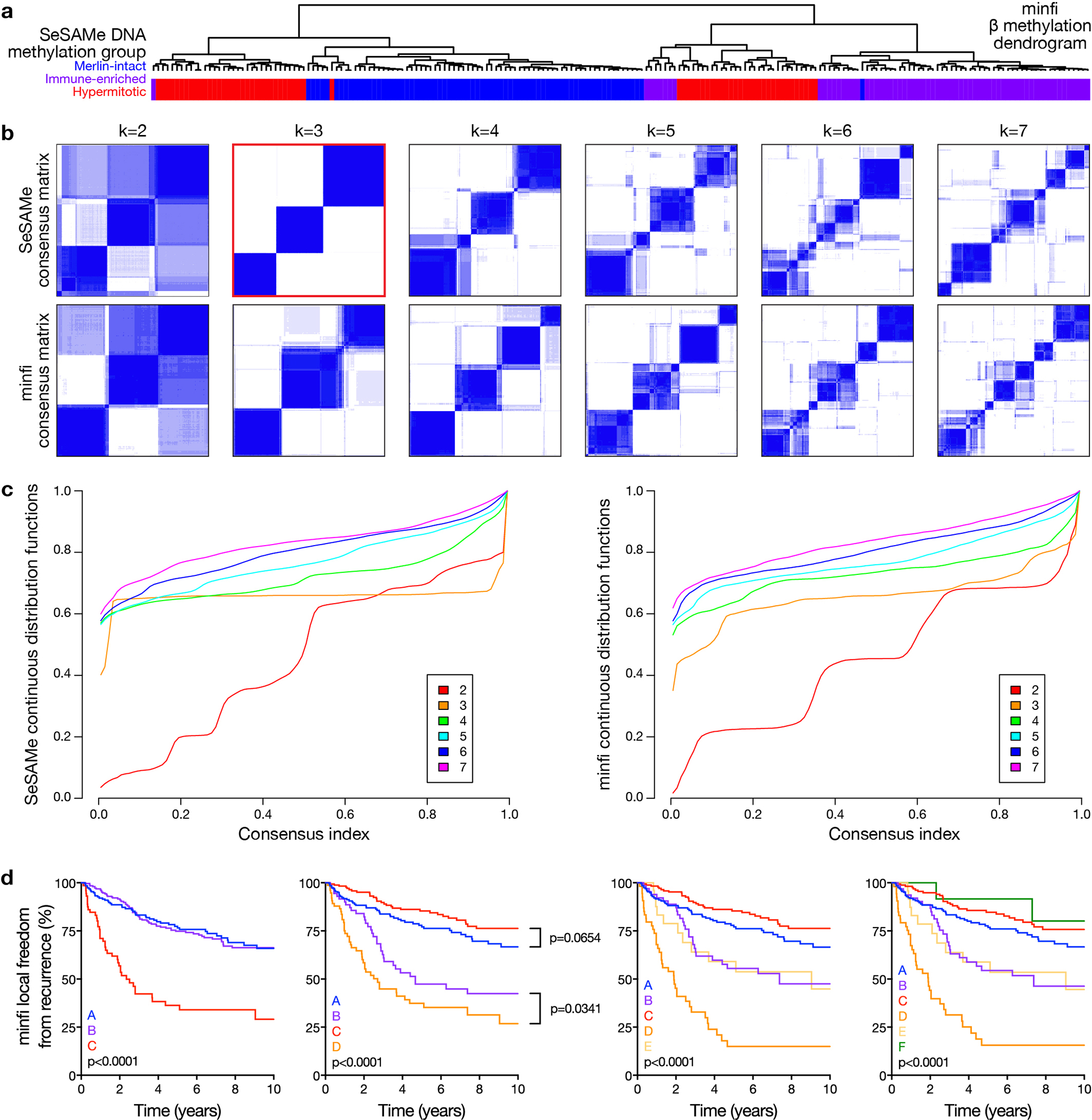
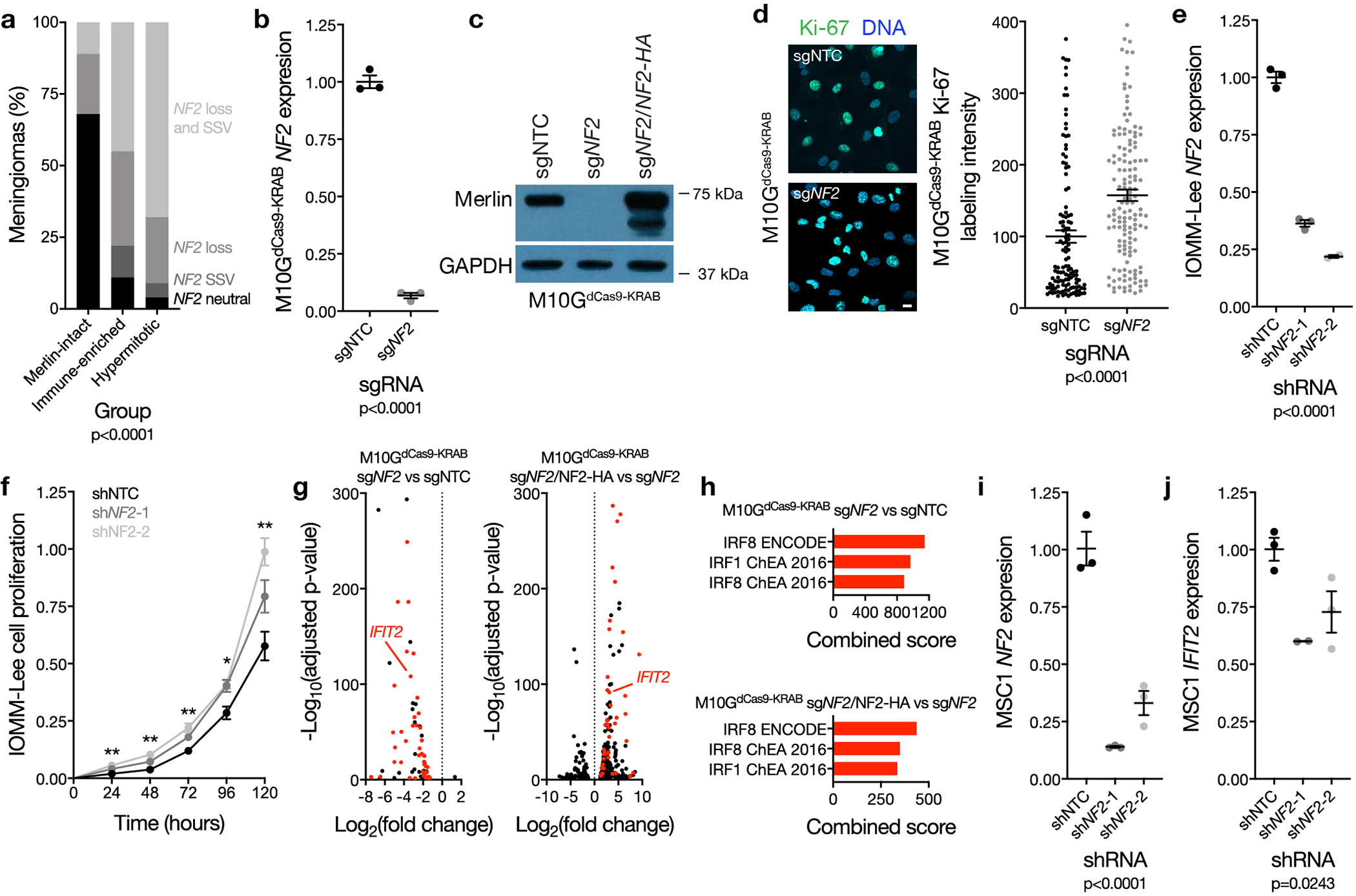
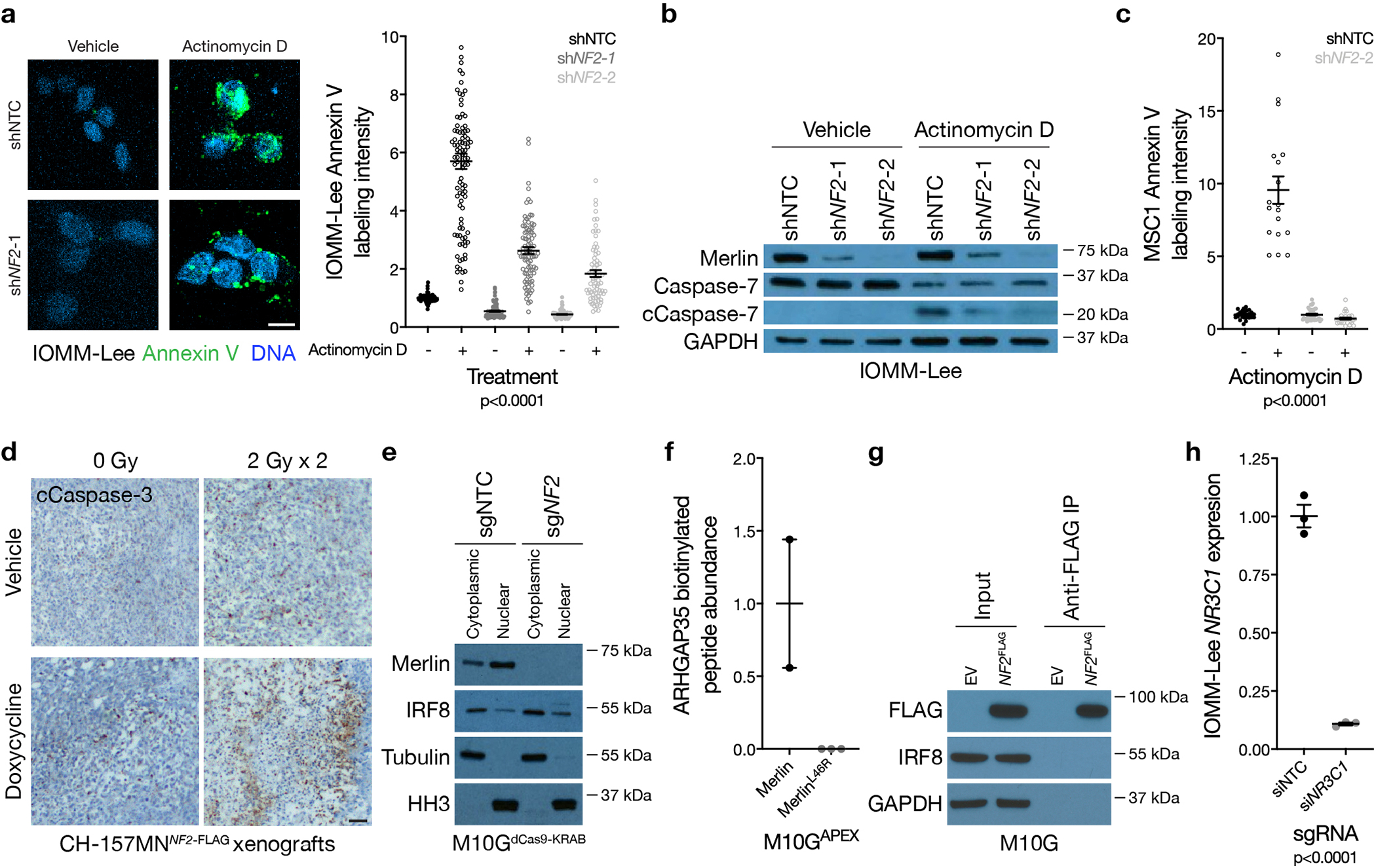
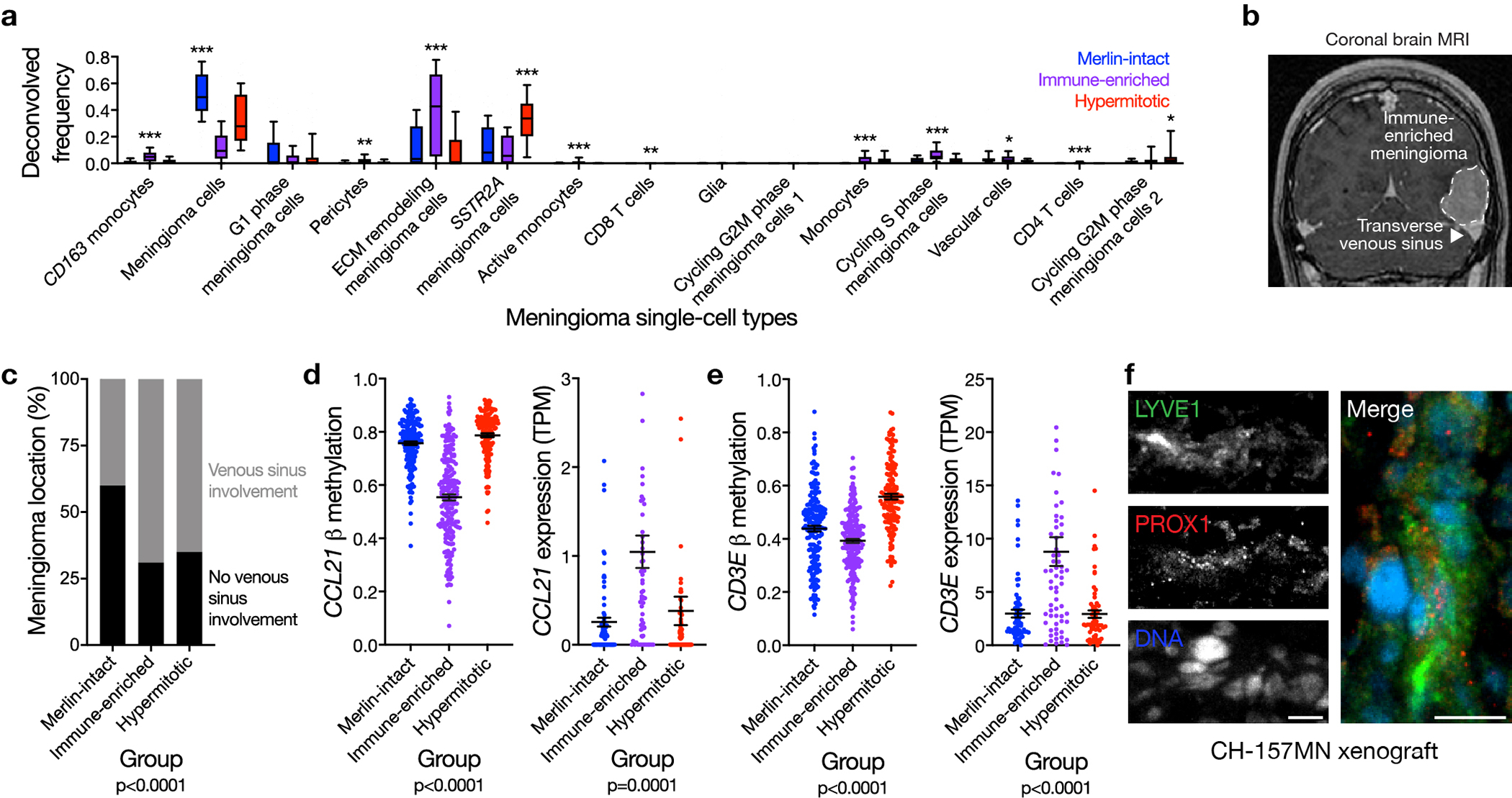
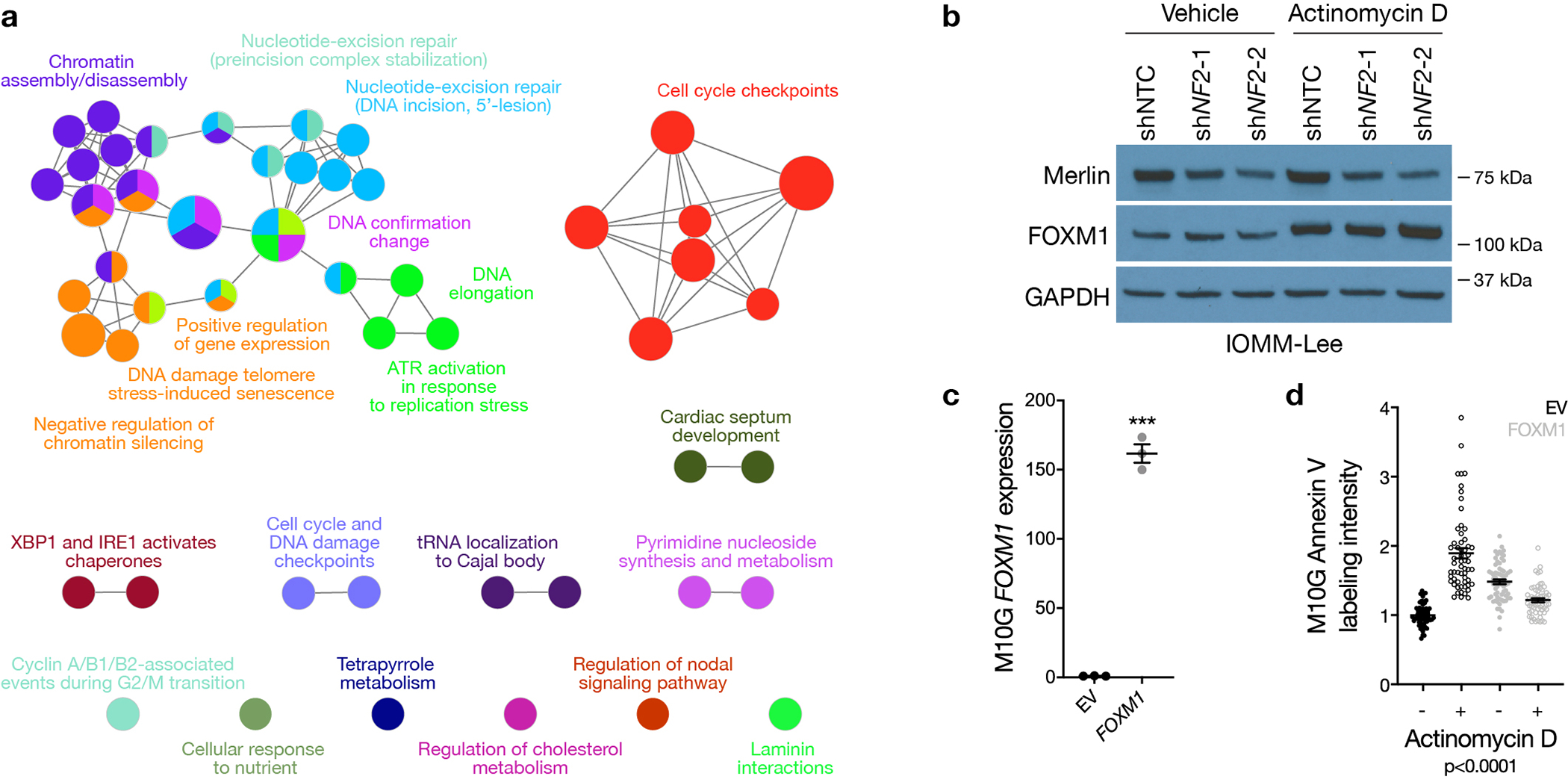
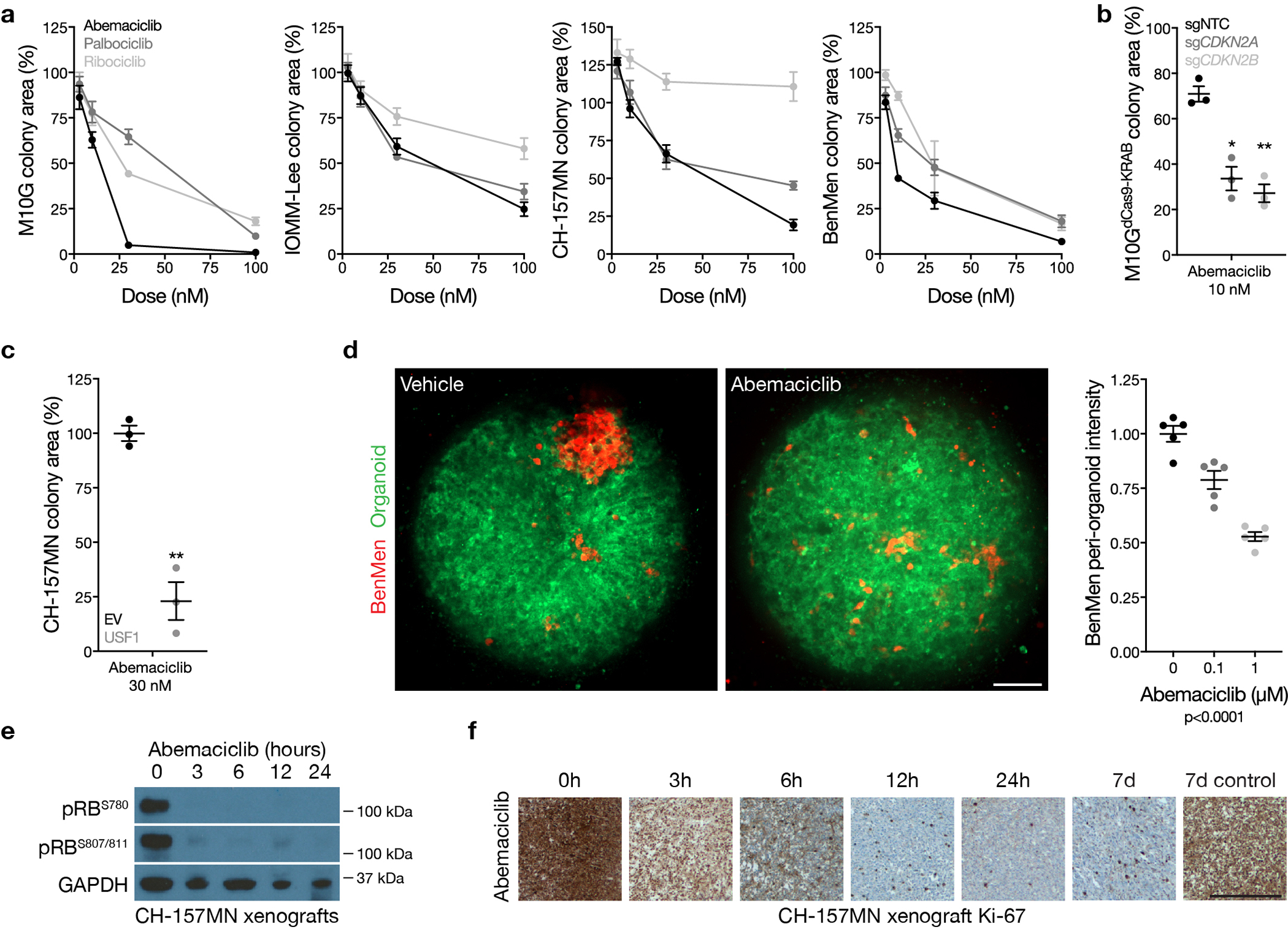
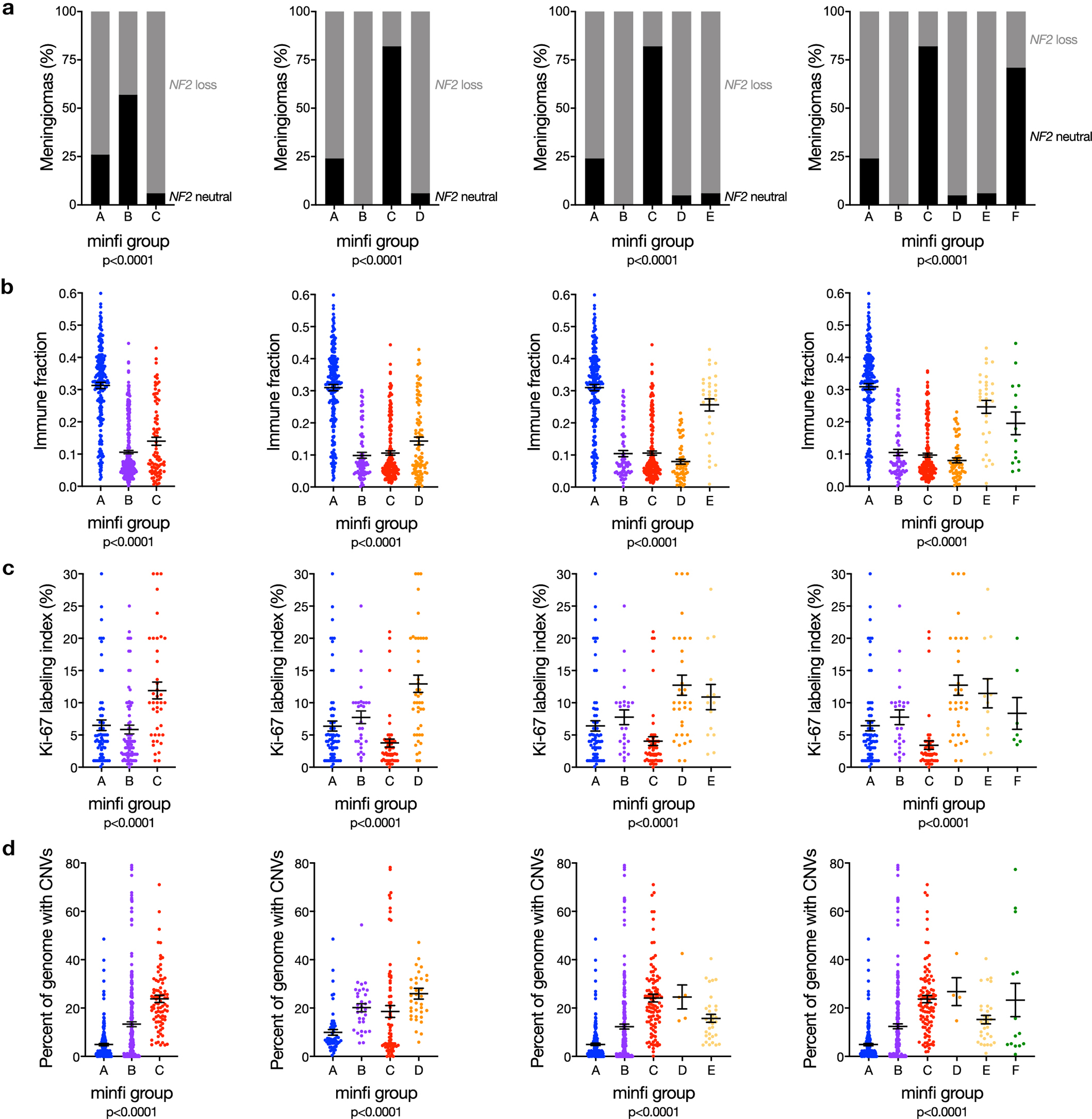
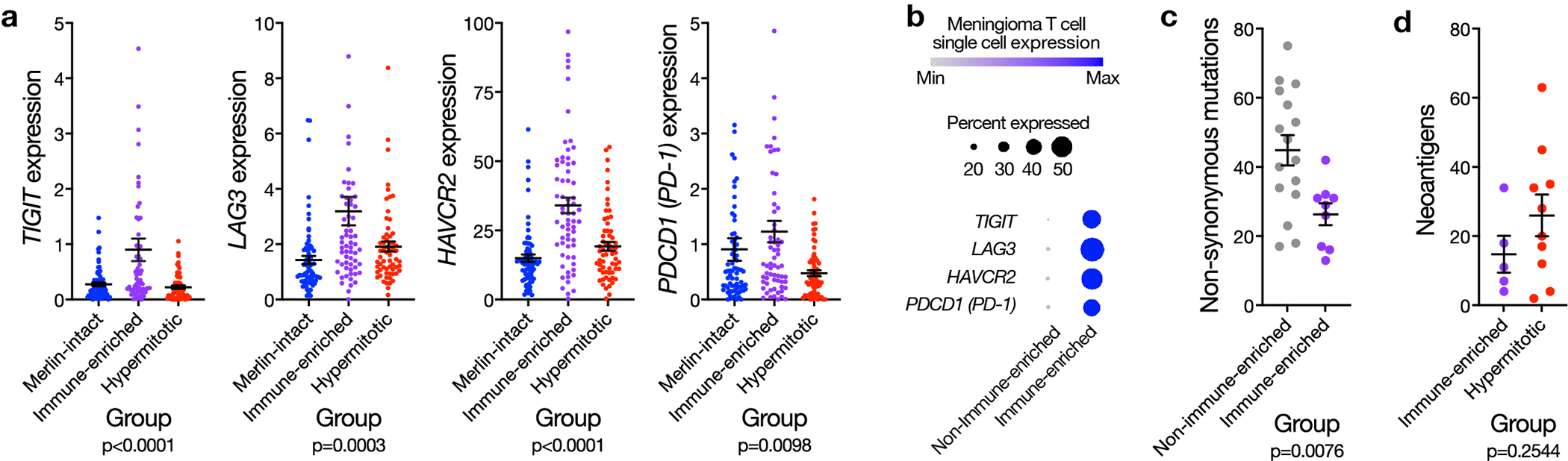
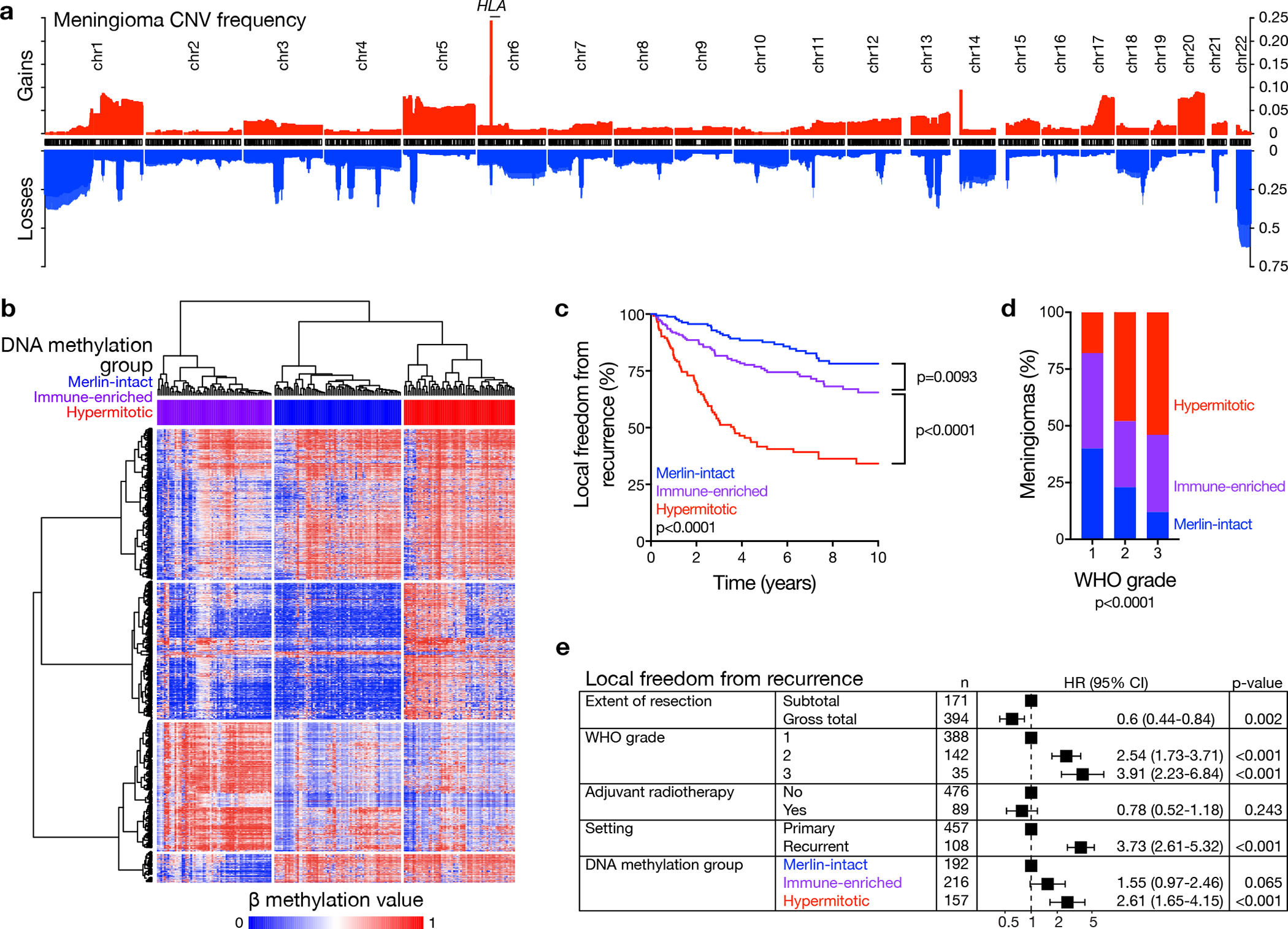
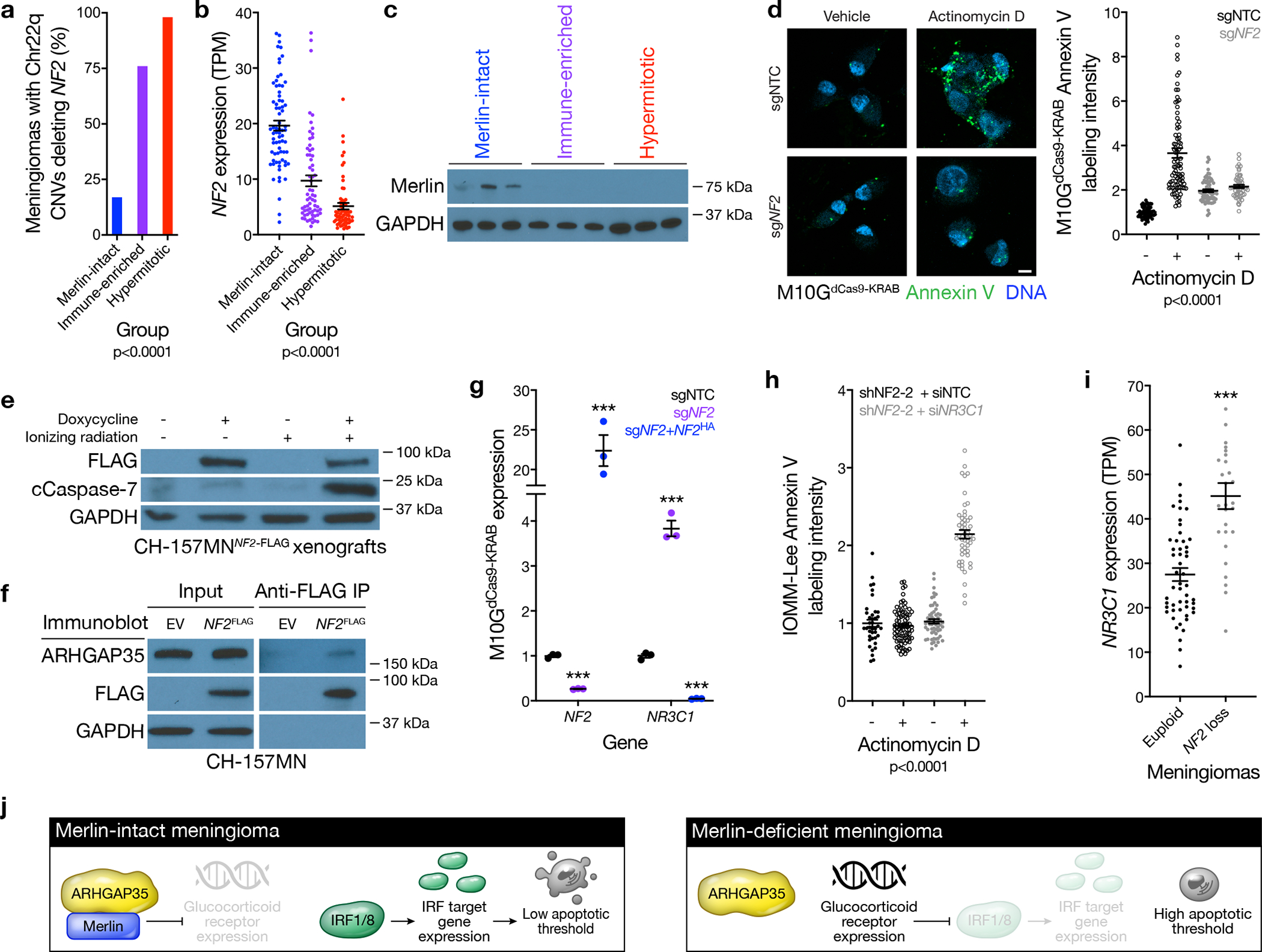
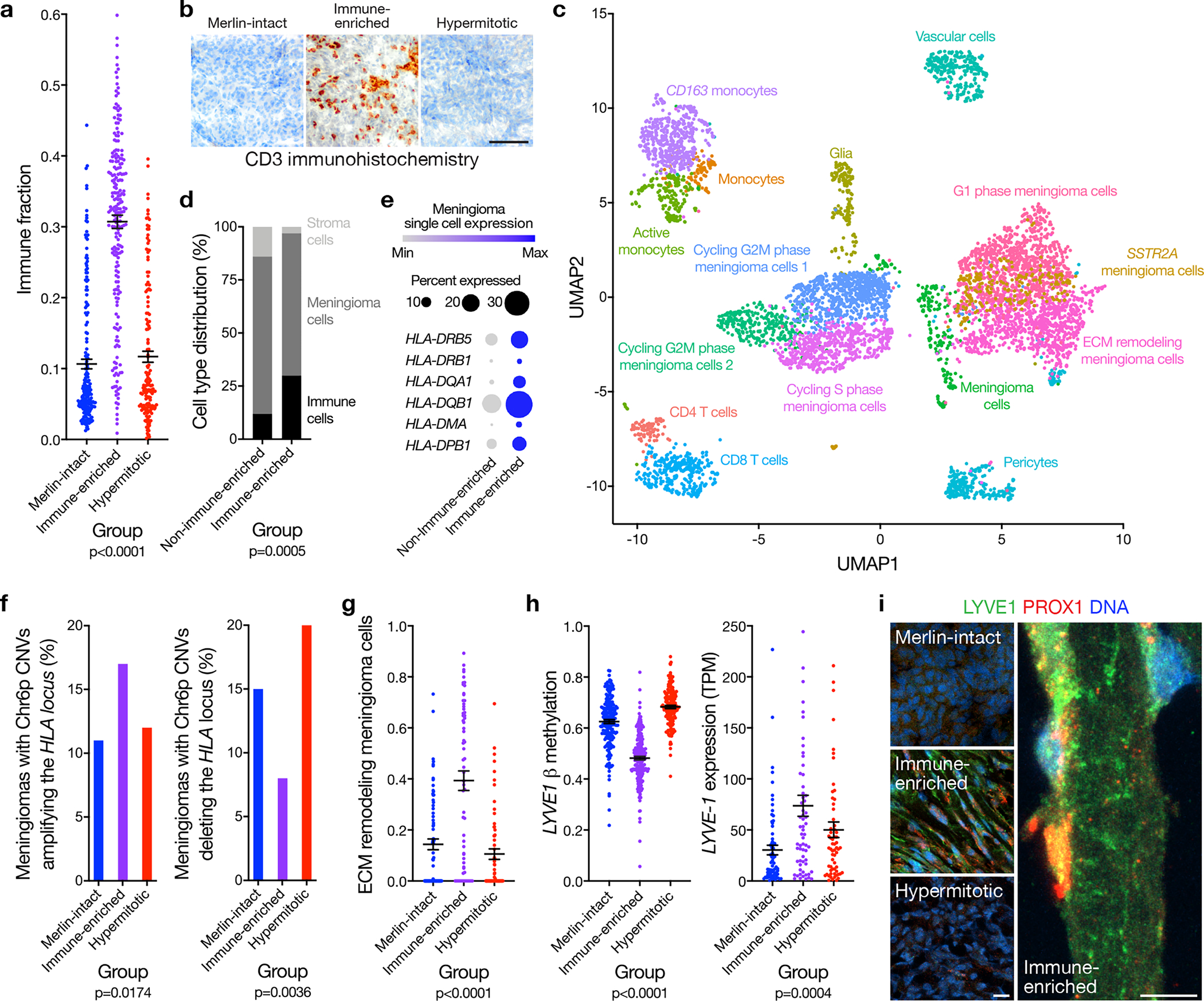
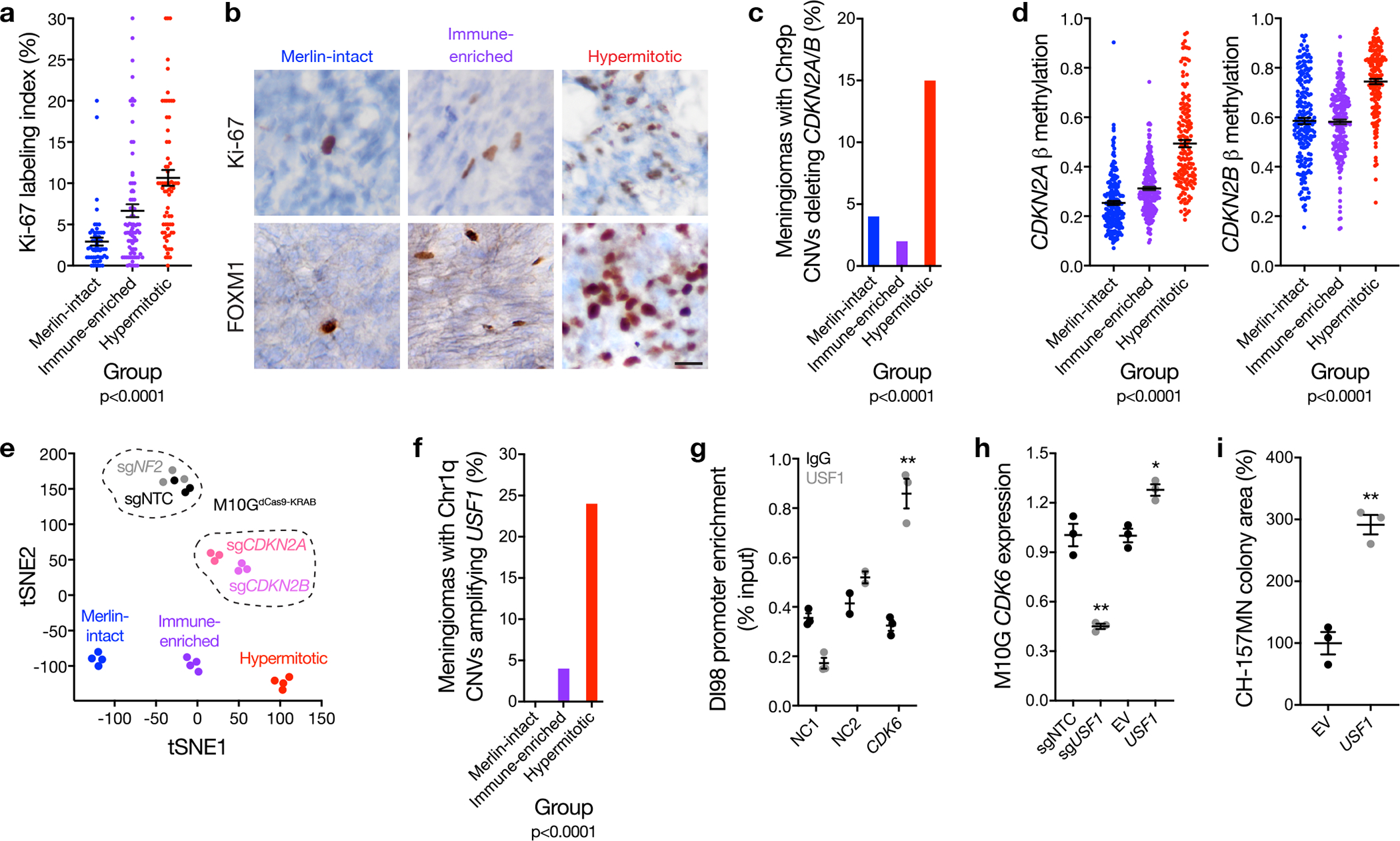
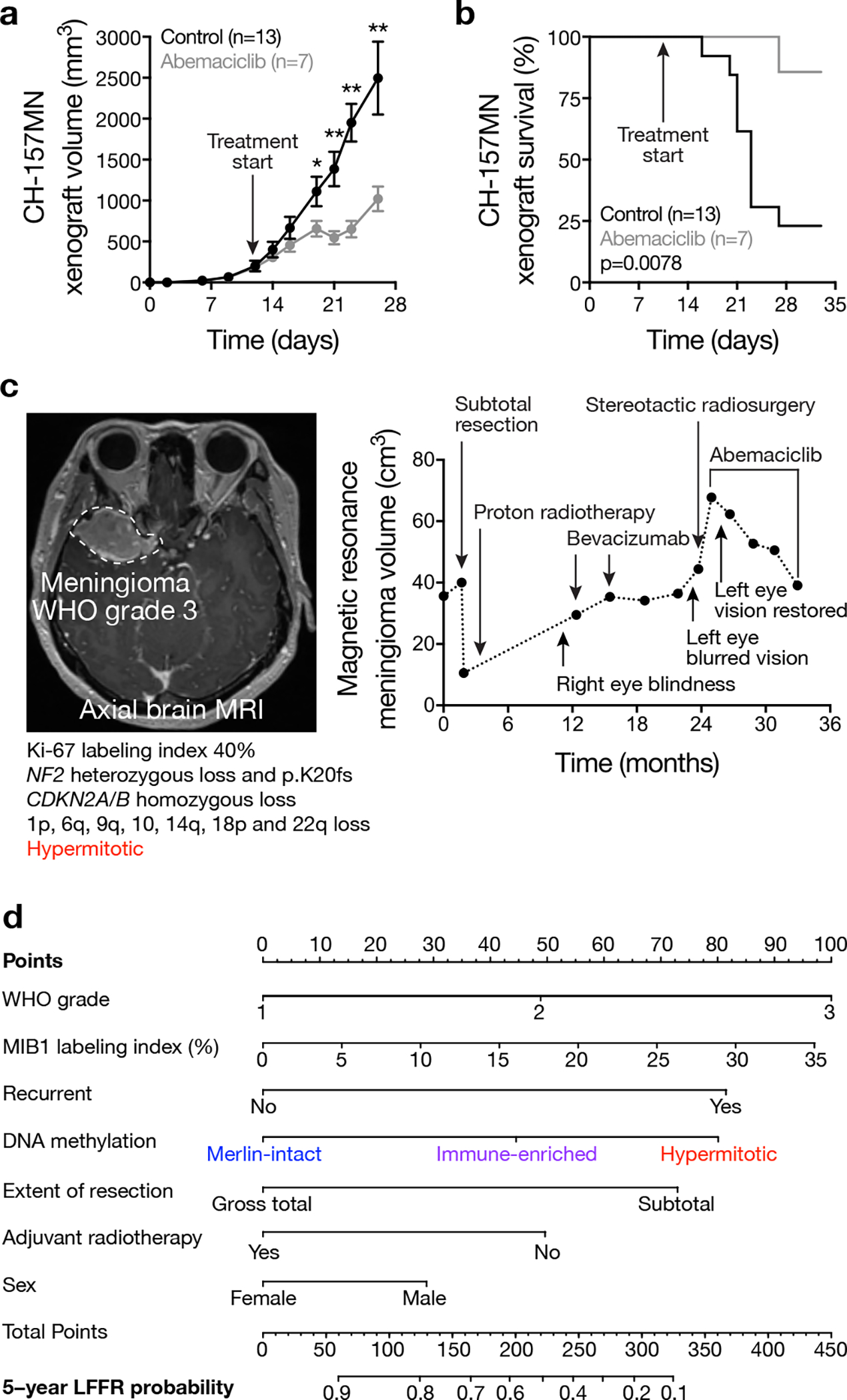
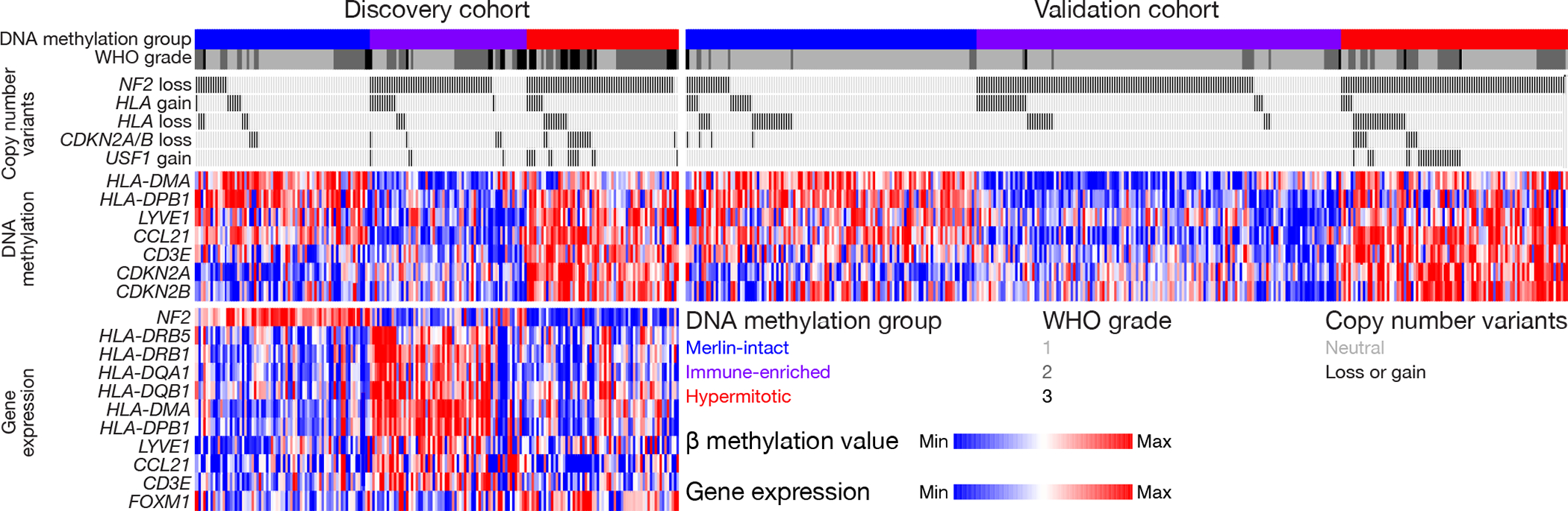
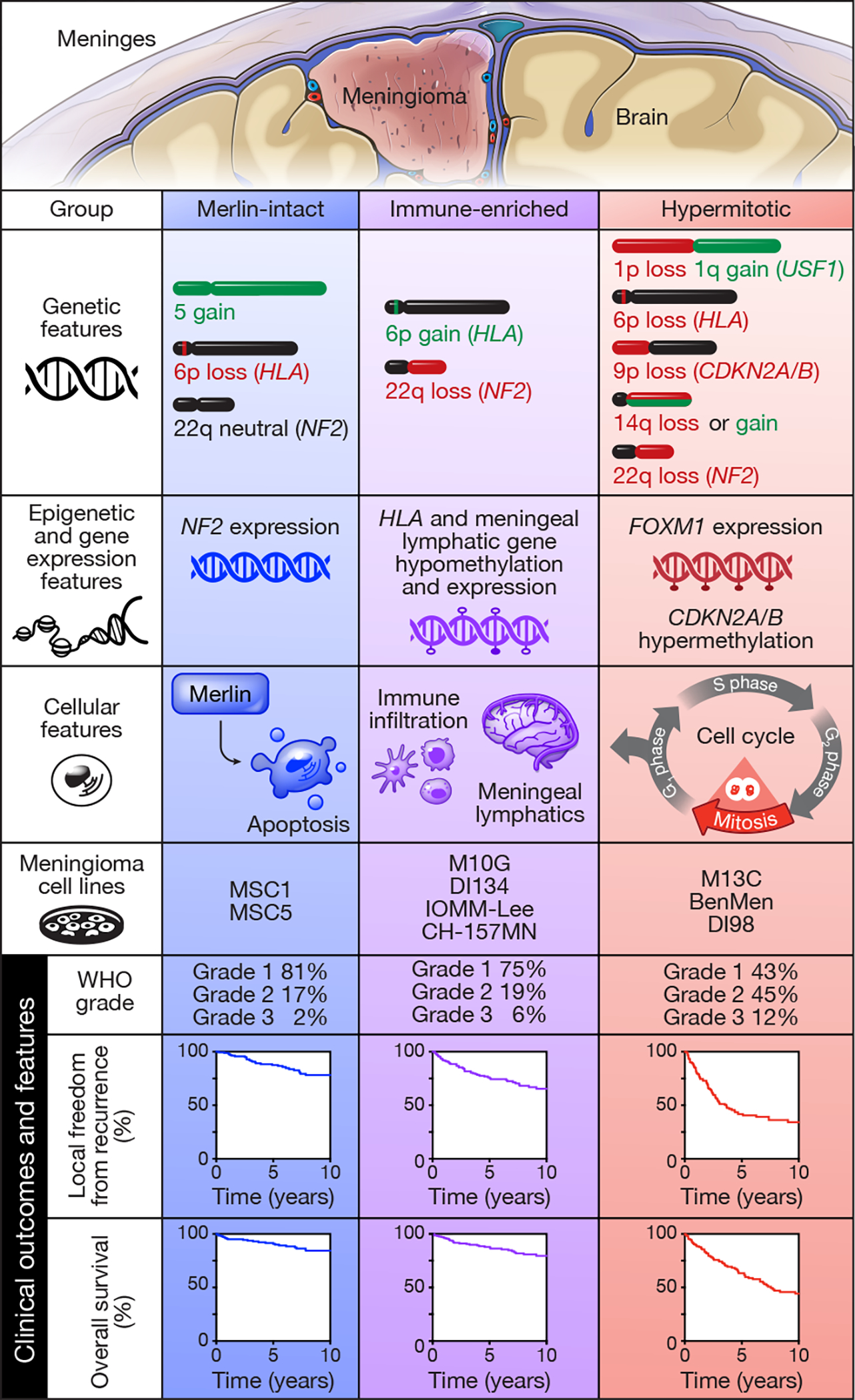
Meningiomas are the most common primary intracranial tumors. There are no effective medical therapies for meningioma patients, and new treatments have been encumbered by limited understanding of meningioma biology. Here, we use DNA methylation profiling on 565 meningiomas integrated with genetic, transcriptomic, biochemical, proteomic and single-cell approaches to show meningiomas are composed of three DNA methylation groups with distinct clinical outcomes, biological drivers and therapeutic vulnerabilities. Merlin-intact meningiomas (34%) have the best outcomes and are distinguished by NF2/Merlin regulation of susceptibility to cytotoxic therapy. Immune-enriched meningiomas (38%) have intermediate outcomes and are distinguished by immune infiltration, HLA expression and lymphatic vessels. Hypermitotic meningiomas (28%) have the worst outcomes and are distinguished by convergent genetic and epigenetic mechanisms driving the cell cycle and resistance to cytotoxic therapy. To translate these findings into clinical practice, we show cytostatic cell cycle inhibitors attenuate meningioma growth in cell culture, organoids, xenografts and patients.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
