

Intestinal epithelial cell metabolism at the interface of microbial dysbiosis and tissue injury
- PMID: 35534699
- PMCID: PMC9259489
- DOI: 10.1038/s41385-022-00514-x
Intestinal epithelial cell metabolism at the interface of microbial dysbiosis and tissue injury
Abstract
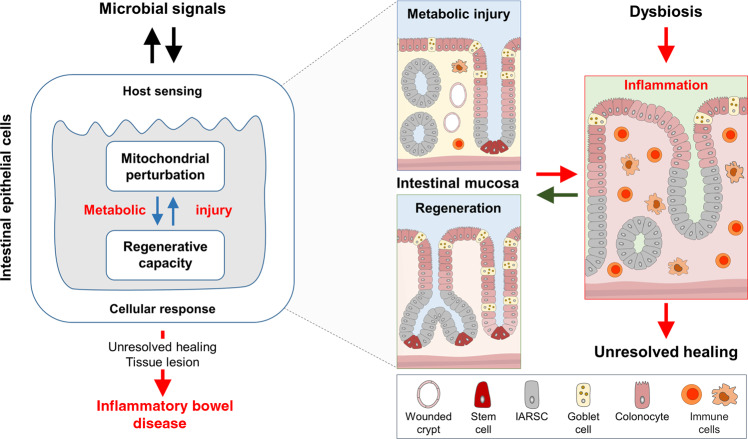
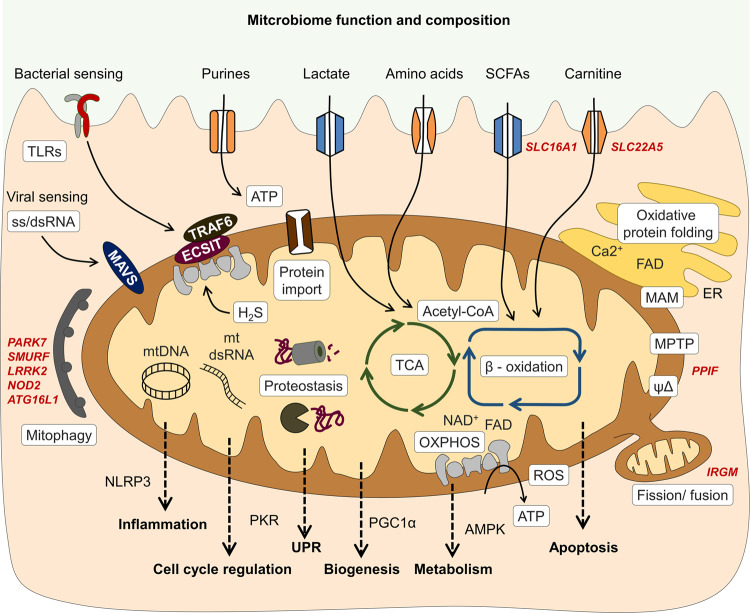
The intestinal epithelium represents the most regenerative tissue in the human body, located in proximity to the dense and functionally diverse microbial milieu of the microbiome. Episodes of tissue injury and incomplete healing of the intestinal epithelium are a prerequisite for immune reactivation and account for recurrent, chronically progressing phenotypes of inflammatory bowel diseases (IBD). Mitochondrial dysfunction and associated changes in intestinal epithelial functions are emerging concepts in the pathogenesis of IBD, suggesting impaired metabolic flexibility of epithelial cells affects the regenerative capacity of the intestinal tissue. Next to rendering the intestinal mucosa susceptible to inflammatory triggers, metabolic reprogramming of the epithelium is implicated in shaping adverse microbial environments. In this review, we introduce the concept of "metabolic injury" as a cell autonomous mechanism of tissue wounding in response to mitochondrial perturbation. Furthermore, we highlight epithelial metabolism as intersection of microbiome, immune cells and epithelial regeneration.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Development, validation and implementation of an in vitro model for the study of metabolic and immune function in normal and inflamed human colonic epithelium.Dan Med J. 2015 Jan;62(1):B4973. Dan Med J. 2015. PMID: 25557335 Review.
-
New Insights into the Role of Oral Microbiota Dysbiosis in the Pathogenesis of Inflammatory Bowel Disease.Dig Dis Sci. 2022 Jan;67(1):42-55. doi: 10.1007/s10620-021-06837-2. Epub 2021 Feb 1. Dig Dis Sci. 2022. PMID: 33527328 Review.
-
Metabolism at the centre of the host-microbe relationship.Clin Exp Immunol. 2019 Aug;197(2):193-204. doi: 10.1111/cei.13329. Epub 2019 Jun 7. Clin Exp Immunol. 2019. PMID: 31107965 Free PMC article. Review.
-
Intestinal microbiota in inflammatory bowel disease: friend of foe?World J Gastroenterol. 2011 Feb 7;17(5):557-66. doi: 10.3748/wjg.v17.i5.557. World J Gastroenterol. 2011. PMID: 21350704 Free PMC article.
-
Dismicrobism in inflammatory bowel disease and colorectal cancer: changes in response of colocytes.World J Gastroenterol. 2014 Dec 28;20(48):18121-30. doi: 10.3748/wjg.v20.i48.18121. World J Gastroenterol. 2014. PMID: 25561781 Free PMC article. Review.
Cited by
-
Special Issue on the "Regulation and Physiopathology of the Gut Barrier".Int J Mol Sci. 2022 Sep 13;23(18):10638. doi: 10.3390/ijms231810638. Int J Mol Sci. 2022. PMID: 36142548 Free PMC article.
-
Inflammation and mitochondria in the pathogenesis of chronic Chagas disease cardiomyopathy.Exp Biol Med (Maywood). 2023 Nov;248(22):2062-2071. doi: 10.1177/15353702231220658. Exp Biol Med (Maywood). 2023. PMID: 38235691 Free PMC article. Review.
-
Gut dysbiosis: Ecological causes and causative effects on human disease.Proc Natl Acad Sci U S A. 2023 Dec 12;120(50):e2316579120. doi: 10.1073/pnas.2316579120. Epub 2023 Dec 4. Proc Natl Acad Sci U S A. 2023. PMID: 38048456 Free PMC article.
-
Dietary xylo-oligosaccharides and arabinoxylans improved growth efficiency by reducing gut epithelial cell turnover in broiler chickens.J Anim Sci Biotechnol. 2024 Mar 4;15(1):35. doi: 10.1186/s40104-024-00991-z. J Anim Sci Biotechnol. 2024. PMID: 38433214 Free PMC article.
-
Effect of Diquat on gut health: molecular mechanisms, toxic effects, and protective strategies.Front Pharmacol. 2025 May 12;16:1562182. doi: 10.3389/fphar.2025.1562182. eCollection 2025. Front Pharmacol. 2025. PMID: 40421207 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
