

The flying spider-monkey tree fern genome provides insights into fern evolution and arborescence
- PMID: 35534720
- PMCID: PMC9122828
- DOI: 10.1038/s41477-022-01146-6
The flying spider-monkey tree fern genome provides insights into fern evolution and arborescence
Erratum in
-
Author Correction: The flying spider-monkey tree fern genome provides insights into fern evolution and arborescence.Nat Plants. 2024 Feb;10(2):344. doi: 10.1038/s41477-024-01631-0. Nat Plants. 2024. PMID: 38307950 Free PMC article. No abstract available.
Abstract
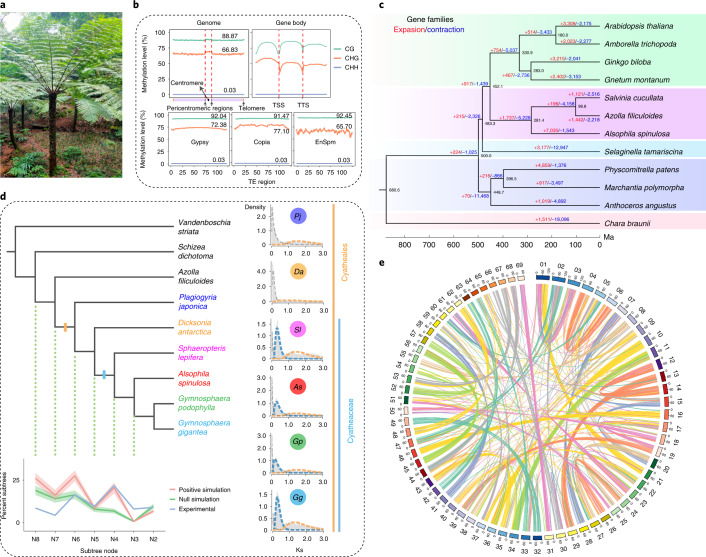
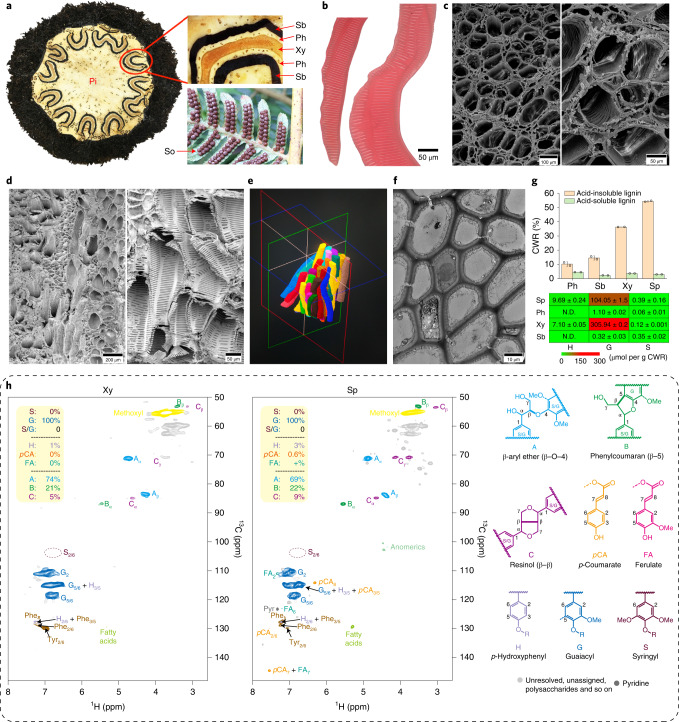
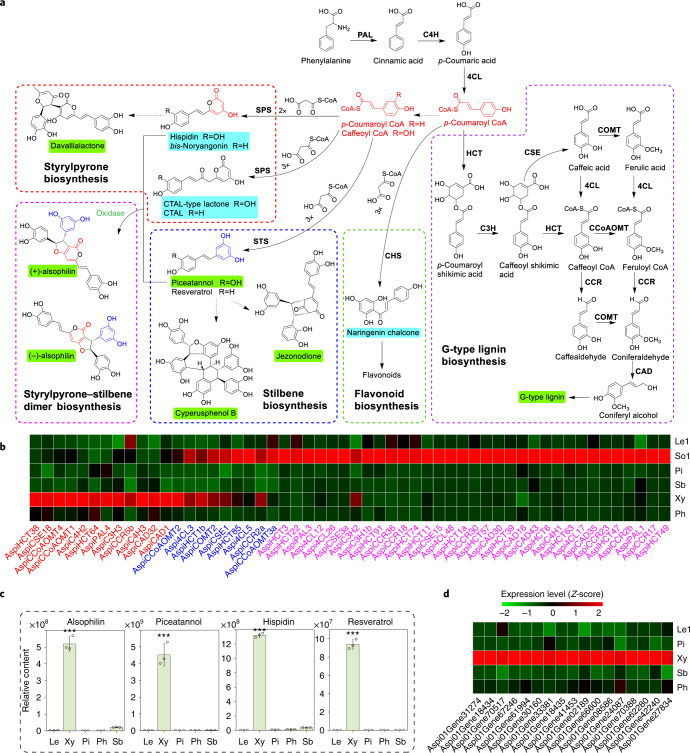
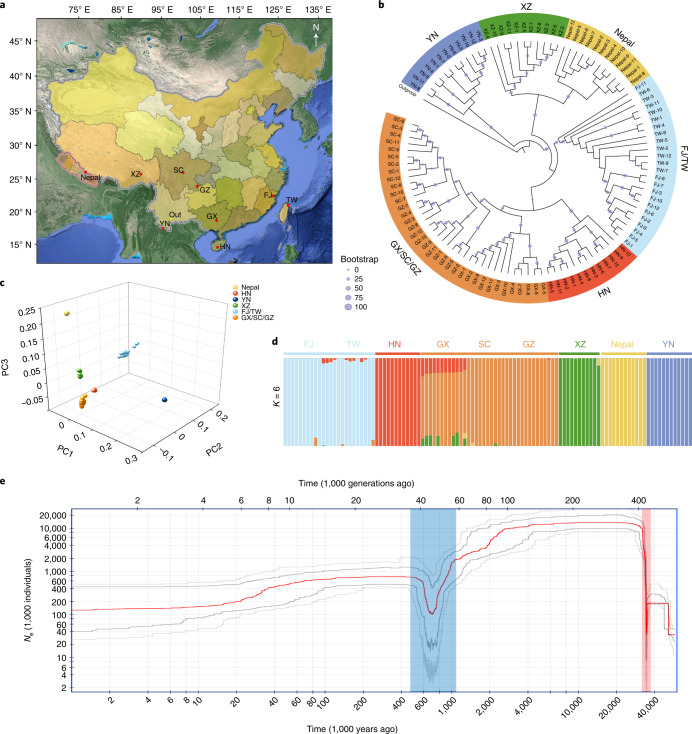
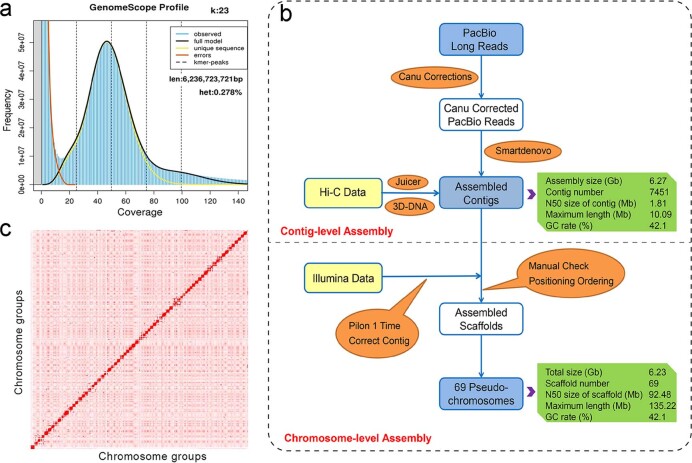
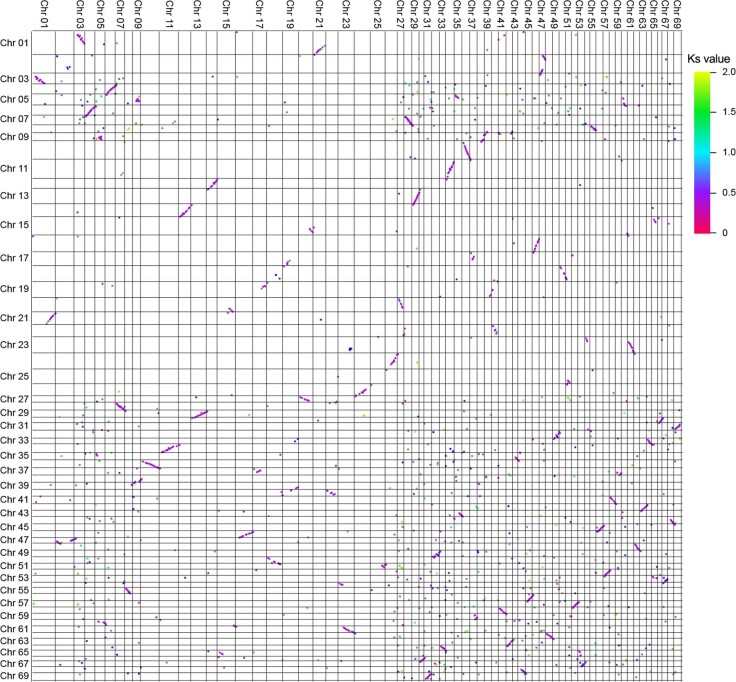
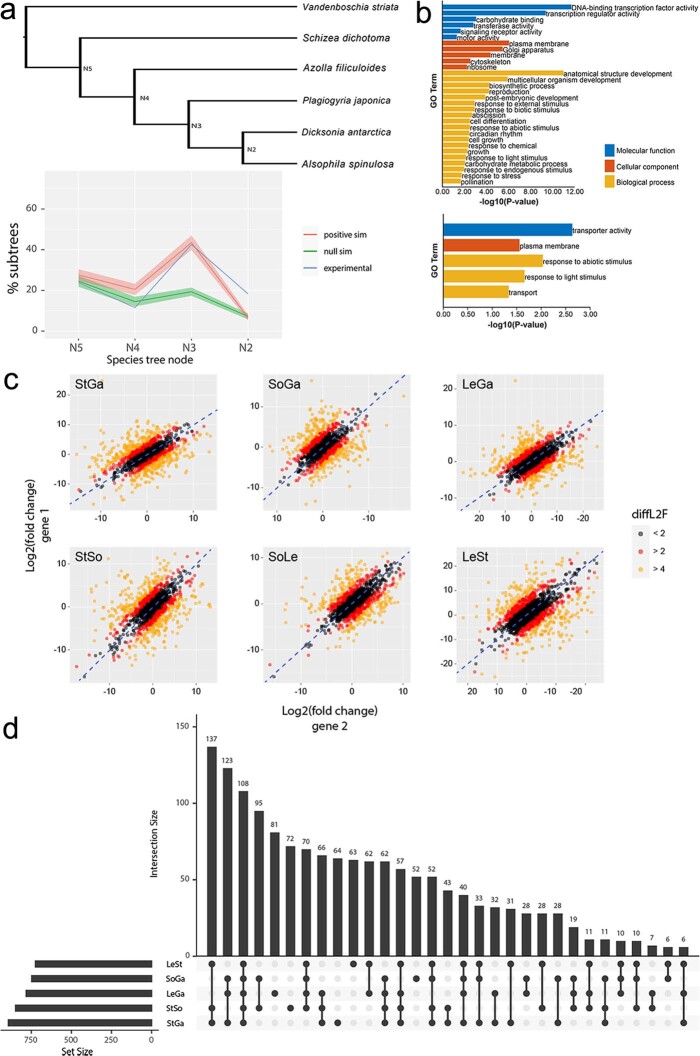
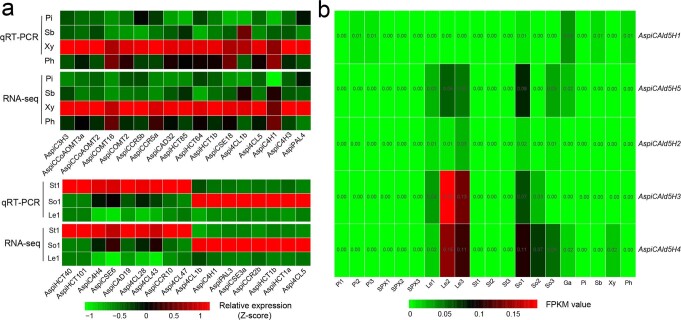
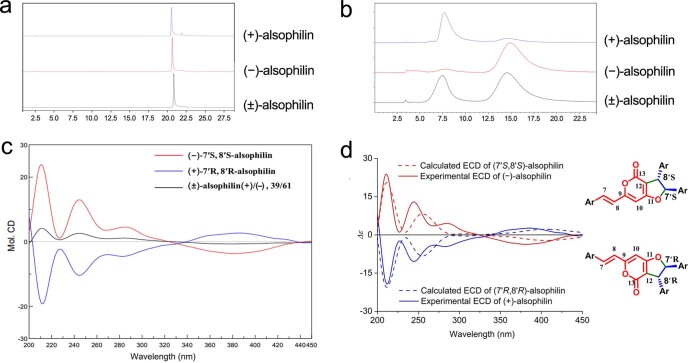
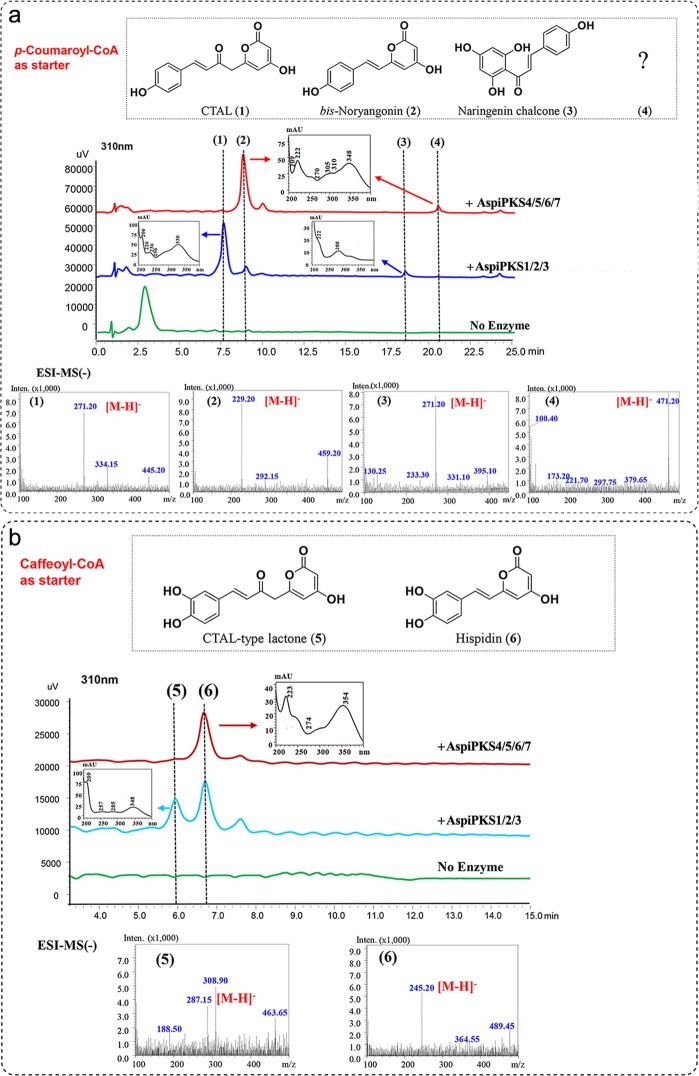
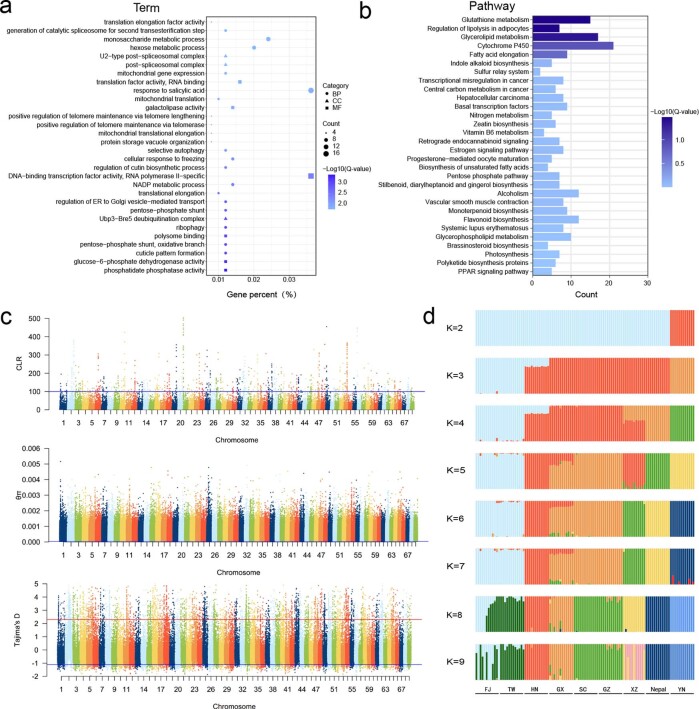
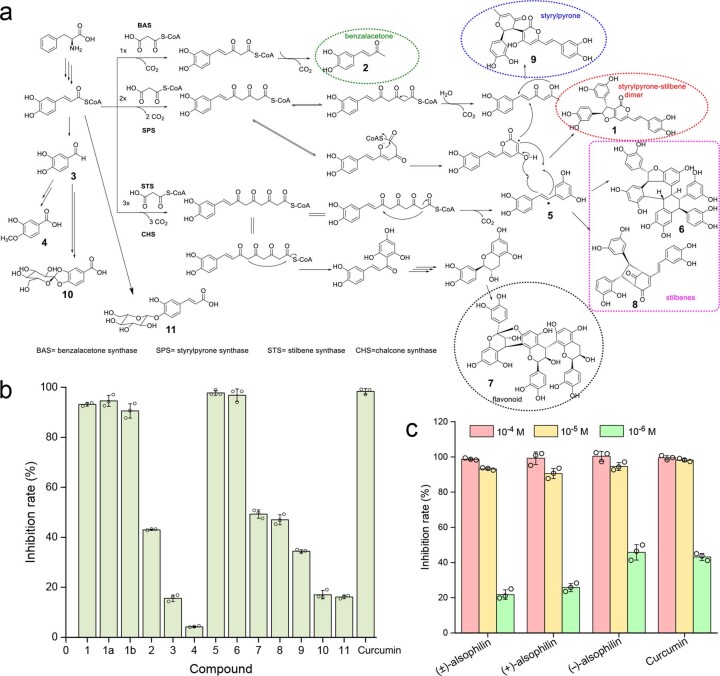
To date, little is known about the evolution of fern genomes, with only two small genomes published from the heterosporous Salviniales. Here we assembled the genome of Alsophila spinulosa, known as the flying spider-monkey tree fern, onto 69 pseudochromosomes. The remarkable preservation of synteny, despite resulting from an ancient whole-genome duplication over 100 million years ago, is unprecedented in plants and probably speaks to the uniqueness of tree ferns. Our detailed investigations into stem anatomy and lignin biosynthesis shed new light on the evolution of stem formation in tree ferns. We identified a phenolic compound, alsophilin, that is abundant in xylem, and we provided the molecular basis for its biosynthesis. Finally, analysis of demographic history revealed two genetic bottlenecks, resulting in rapid demographic declines of A. spinulosa. The A. spinulosa genome fills a crucial gap in the plant genomic landscape and helps elucidate many unique aspects of tree fern biology.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Sarkanen, K. V. & Ludwig, C. H. Lignins: Occurrence, Formation, Structure and Reactions (Wiley-Interscience, 1971).
-
- Schuettpelz E, Schneider H, Smith AR, Kessler M. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 2016;54:563–603. doi: 10.1111/jse.12229. - DOI
-
- Dong SY, Zuo ZY. On the recognition of Gymnosphaera as a distinct genus in Cyatheaceae. Ann. Mo. Bot. Gard. 2018;103:1–23. doi: 10.3417/2017049. - DOI
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
