

An integrative in-silico analysis discloses a novel molecular subset of colorectal cancer possibly eligible for immune checkpoint immunotherapy
- PMID: 35534873
- PMCID: PMC9082922
- DOI: 10.1186/s13062-022-00324-y
An integrative in-silico analysis discloses a novel molecular subset of colorectal cancer possibly eligible for immune checkpoint immunotherapy
Abstract
Background: Historically, the molecular classification of colorectal cancer (CRC) was based on the global genomic status, which identified microsatellite instability in mismatch repair (MMR) deficient CRC, and chromosomal instability in MMR proficient CRC. With the introduction of immune checkpoint inhibitors, the microsatellite and chromosomal instability classification regained momentum as the microsatellite instability condition predicted sensitivity to immune checkpoint inhibitors, possibly due to both high tumor mutation burden (TMB) and high levels of infiltrating lymphocytes. Conversely, proficient MMR CRC are mostly resistant to immunotherapy. To better understand the relationship between the microsatellite and chromosomal instability classification, and eventually discover additional CRC subgroups relevant for therapeutic decisions, we developed a computational pipeline that include molecular integrative analysis of genomic, epigenomic and transcriptomic data.
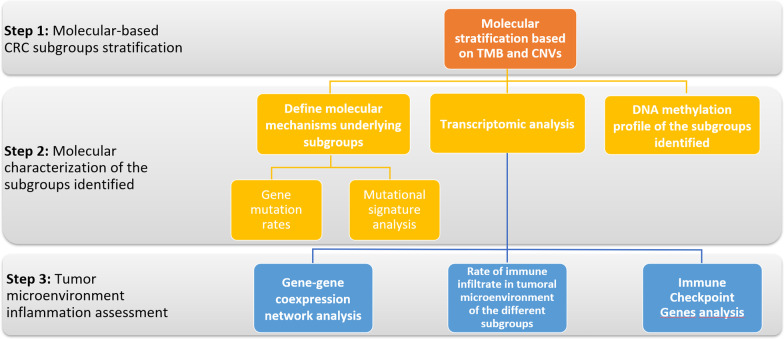
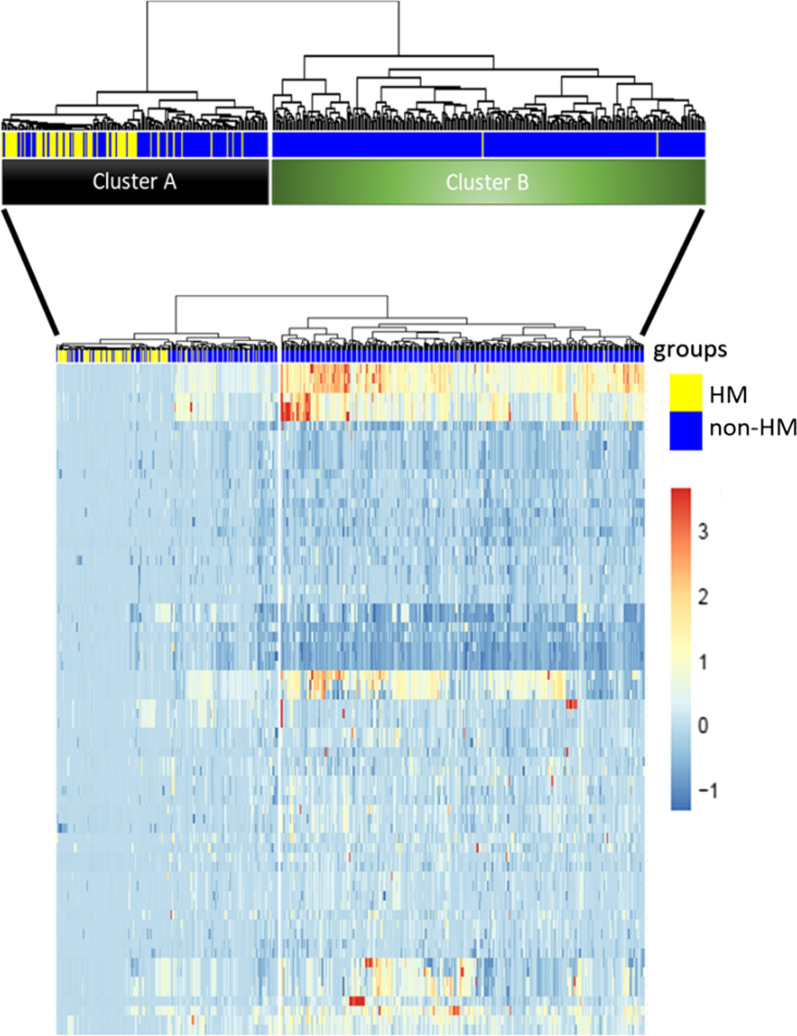
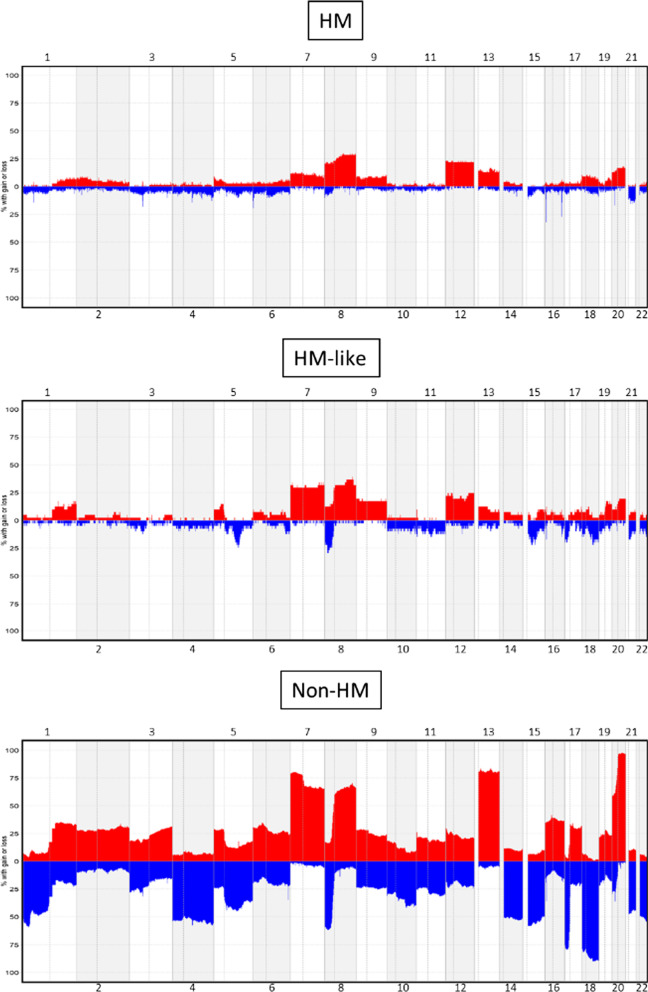
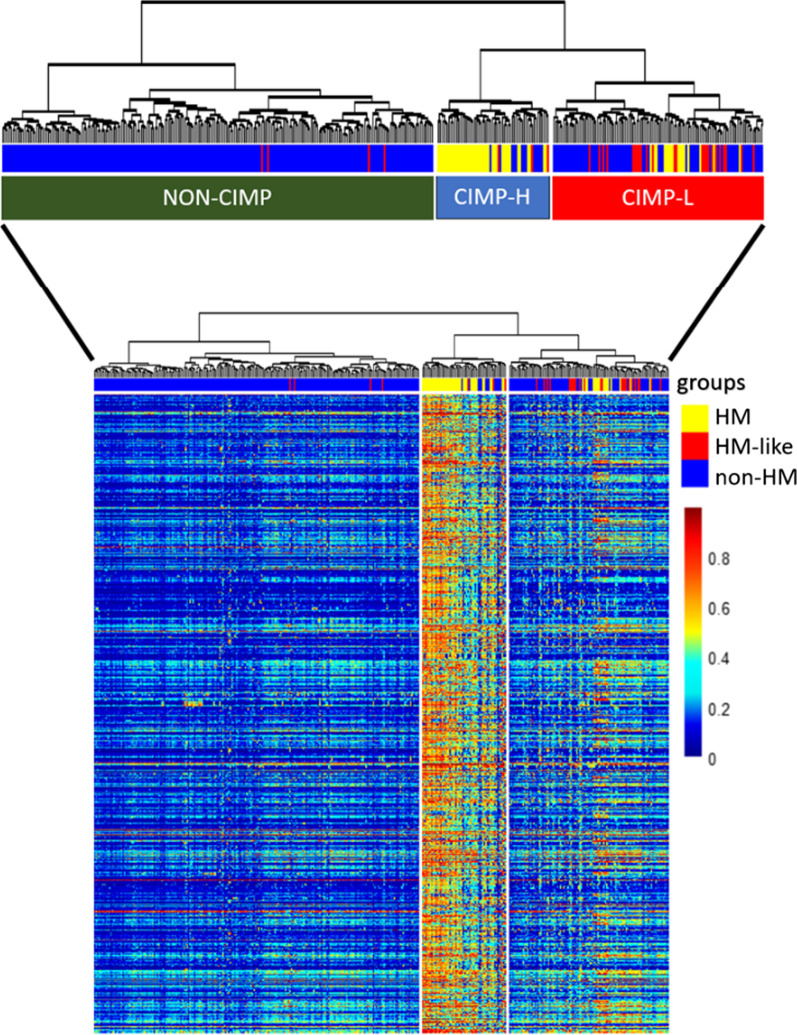
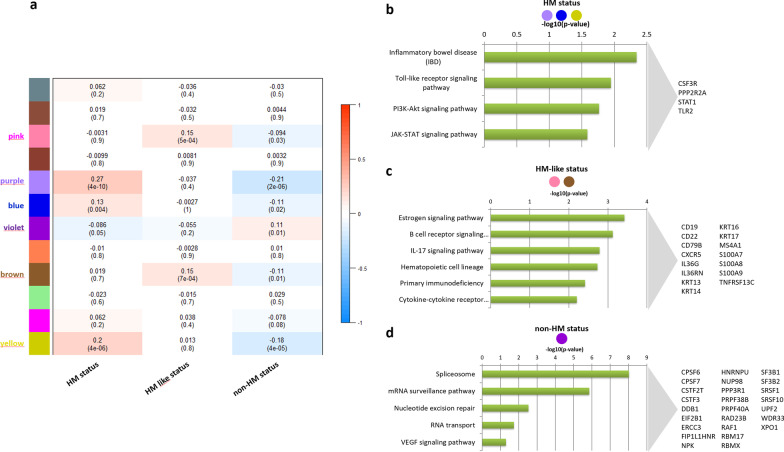
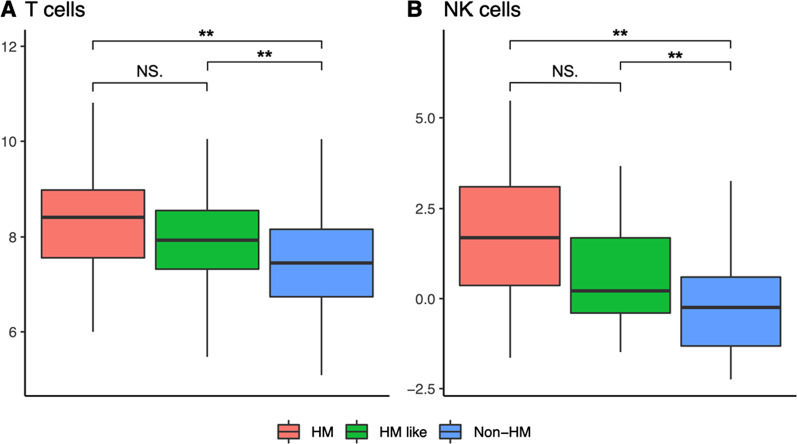
Results: The first step of the pipeline was based on unsupervised hierarchical clustering analysis of copy number variations (CNVs) versus hypermutation status that identified a first CRC cluster with few CNVs enriched in Hypermutated and microsatellite instability samples, a second CRC cluster with a high number of CNVs mostly including non-HM and microsatellite stable samples, and a third cluster (7.8% of the entire dataset) with low CNVs and low TMB, which shared clinical-pathological features with Hypermutated CRCs and thus defined Hypermutated-like CRCs. The mutational features, DNA methylation profile and base substitution fingerprints of these tumors revealed that Hypermutated-like patients are molecularly distinct from Hypermutated and non-Hypermutated tumors and are likely to develop and progress through different genetic events. Transcriptomic analysis highlighted further differences amongst the three groups and revealed an inflamed tumor microenvironment and modulation Immune Checkpoint Genes in Hypermutated-like CRCs.
Conclusion: Therefore, our work highlights Hypermutated-like tumors as a distinct and previously unidentified CRC subgroup possibly responsive to immune checkpoint inhibitors. If further validated, these findings can lead to expanding the fraction of patients eligible to immunotherapy.
Keywords: Colorectal cancer; Immunoinformatics; Immunotherapy; Meta-analysis; Multi-omics.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Cancer Statistics, 2021—PubMed. https://pubmed-ncbi-nlm-nih-gov.ezproxy.uniroma1.it/33433946/. Accessed 13 Dec 2021.
-
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
