

Delineating the RAS Conformational Landscape
- PMID: 35536216
- PMCID: PMC9256797
- DOI: 10.1158/0008-5472.CAN-22-0804
Delineating the RAS Conformational Landscape
Abstract
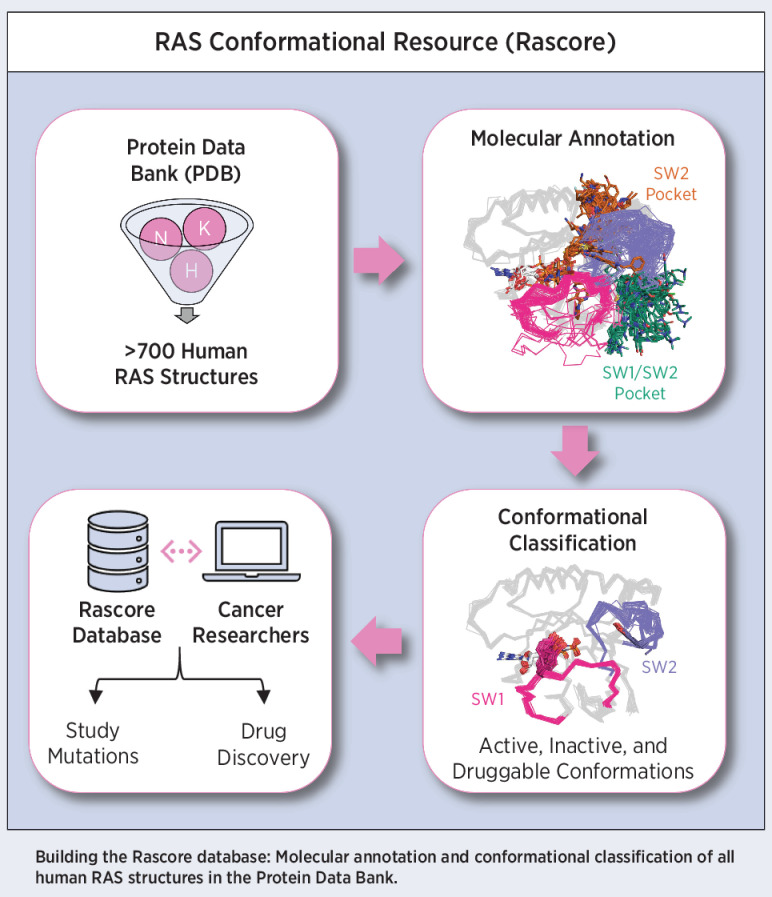
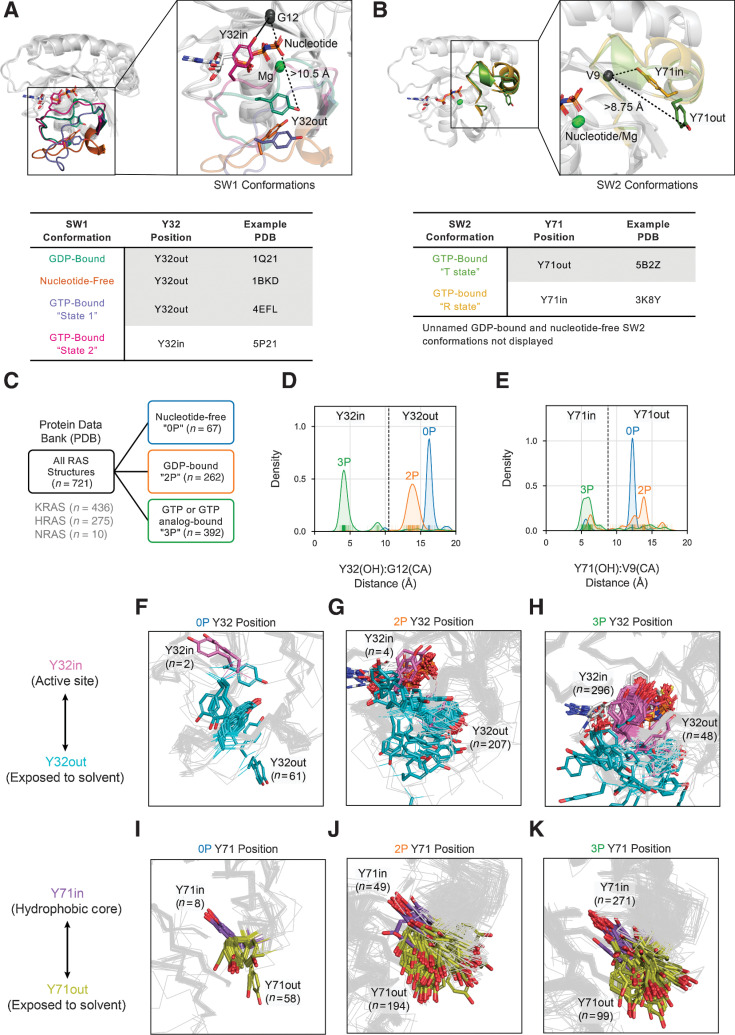
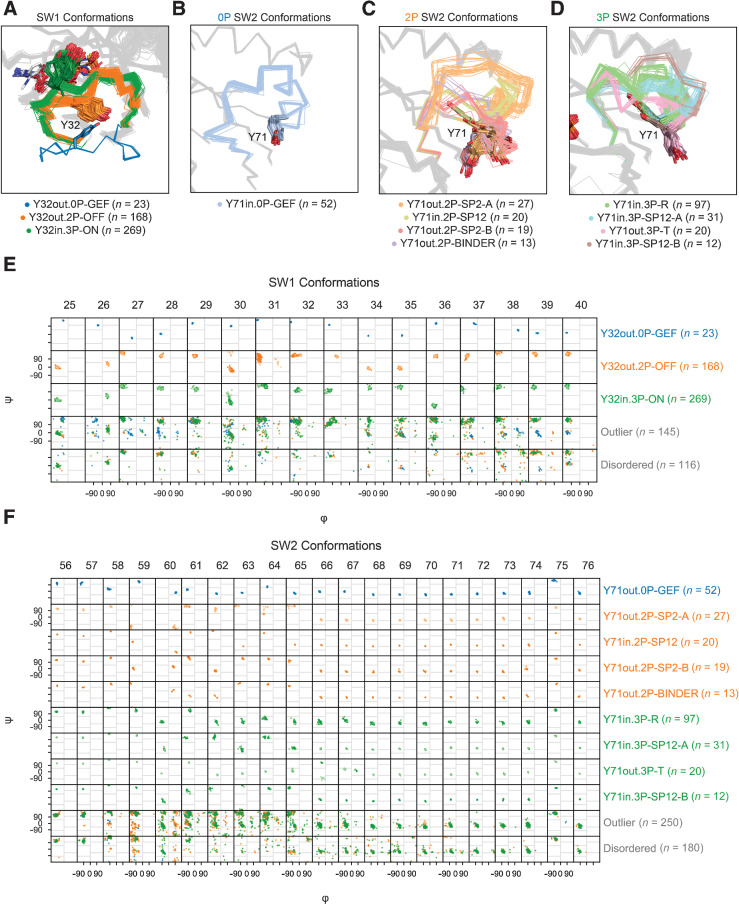
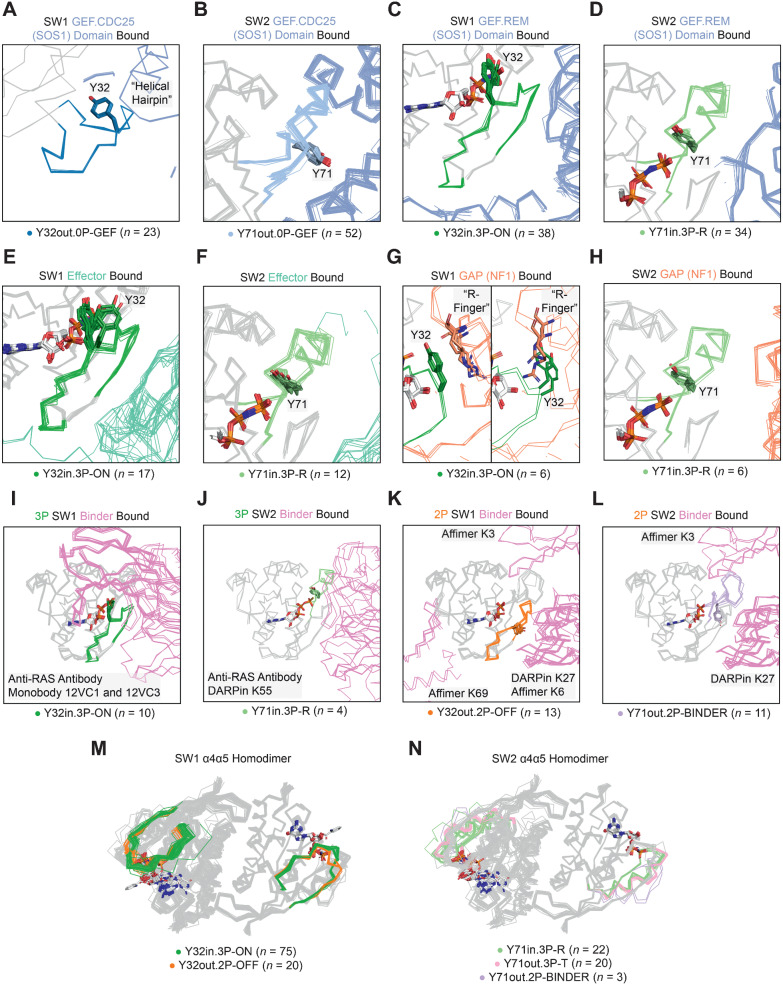
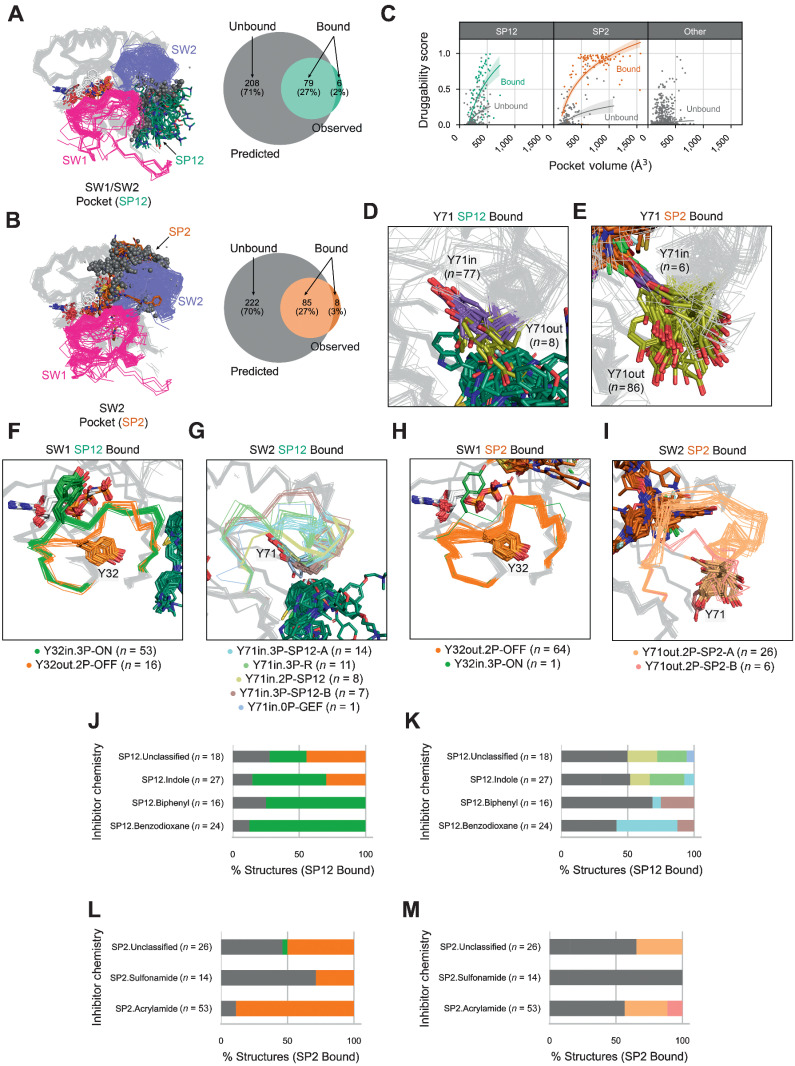
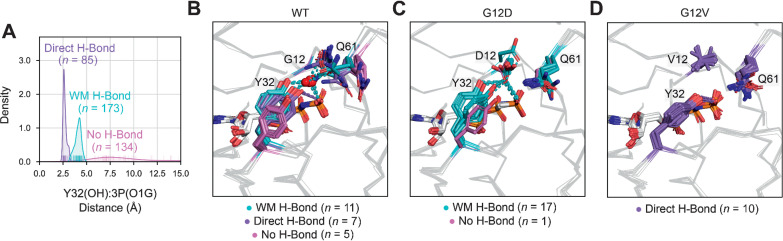
Mutations in RAS isoforms (KRAS, NRAS, and HRAS) are among the most frequent oncogenic alterations in many cancers, making these proteins high priority therapeutic targets. Effectively targeting RAS isoforms requires an exact understanding of their active, inactive, and druggable conformations. However, there is no structural catalog of RAS conformations to guide therapeutic targeting or examining the structural impact of RAS mutations. Here we present an expanded classification of RAS conformations based on analyses of the catalytic switch 1 (SW1) and switch 2 (SW2) loops. From 721 human KRAS, NRAS, and HRAS structures available in the Protein Data Bank (206 RAS-protein cocomplexes, 190 inhibitor-bound, and 325 unbound, including 204 WT and 517 mutated structures), we created a broad conformational classification based on the spatial positions of Y32 in SW1 and Y71 in SW2. Clustering all well-modeled SW1 and SW2 loops using a density-based machine learning algorithm defined additional conformational subsets, some previously undescribed. Three SW1 conformations and nine SW2 conformations were identified, each associated with different nucleotide states (GTP-bound, nucleotide-free, and GDP-bound) and specific bound proteins or inhibitor sites. The GTP-bound SW1 conformation could be further subdivided on the basis of the hydrogen bond type made between Y32 and the GTP γ-phosphate. Further analysis clarified the catalytic impact of G12D and G12V mutations and the inhibitor chemistries that bind to each druggable RAS conformation. Overall, this study has expanded our understanding of RAS structural biology, which could facilitate future RAS drug discovery.
Significance: Analysis of >700 RAS structures helps define an expanded landscape of active, inactive, and druggable RAS conformations, the structural impact of common RAS mutations, and previously uncharacterized RAS inhibitor-binding modes.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, et al. Ras conformational ensembles, allostery, and signaling. Chem Rev 2016;116:6607–65. - PubMed
-
- Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature 1998;394:337–43. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
