

DEMO2: Assemble multi-domain protein structures by coupling analogous template alignments with deep-learning inter-domain restraint prediction
- PMID: 35536281
- PMCID: PMC9252800
- DOI: 10.1093/nar/gkac340
DEMO2: Assemble multi-domain protein structures by coupling analogous template alignments with deep-learning inter-domain restraint prediction
Abstract
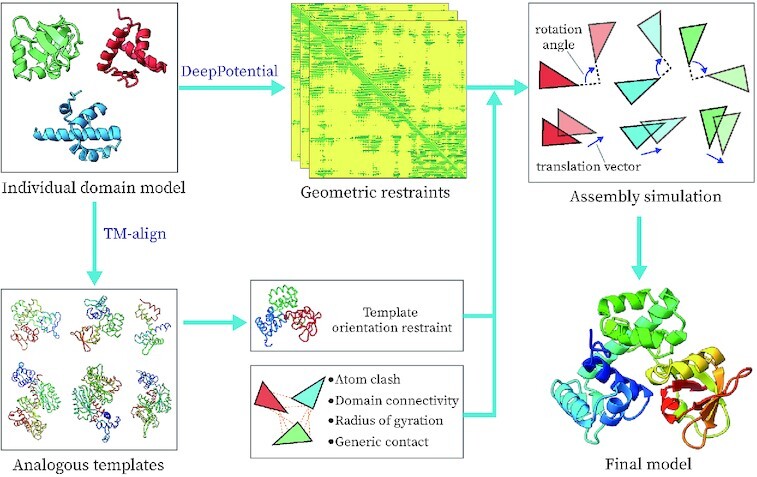
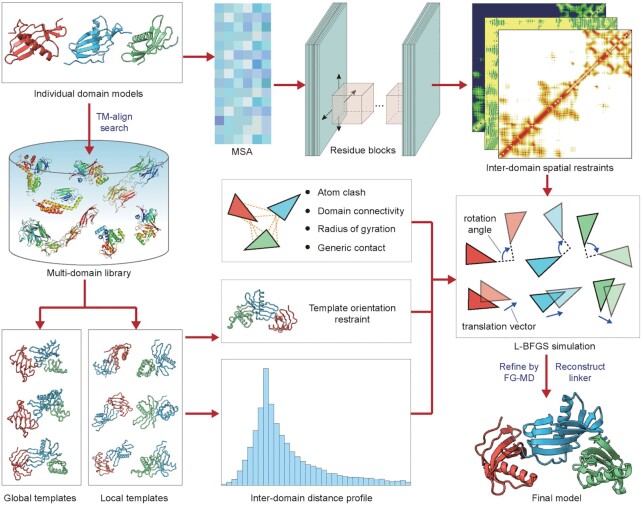
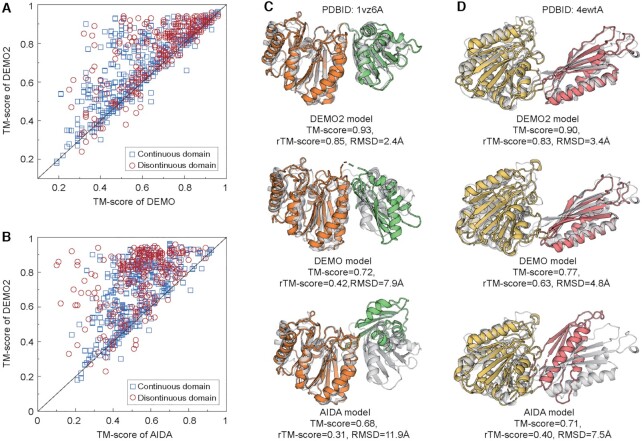
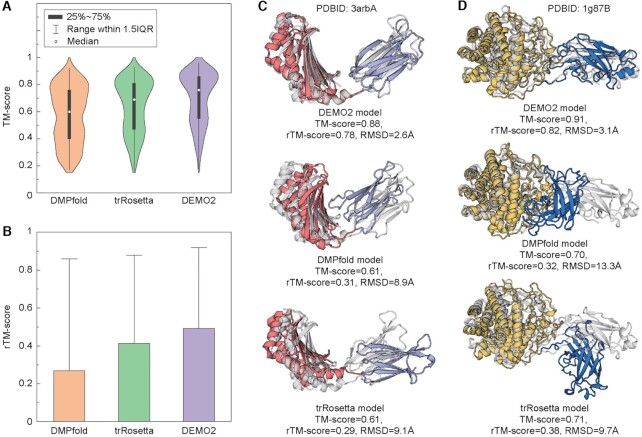
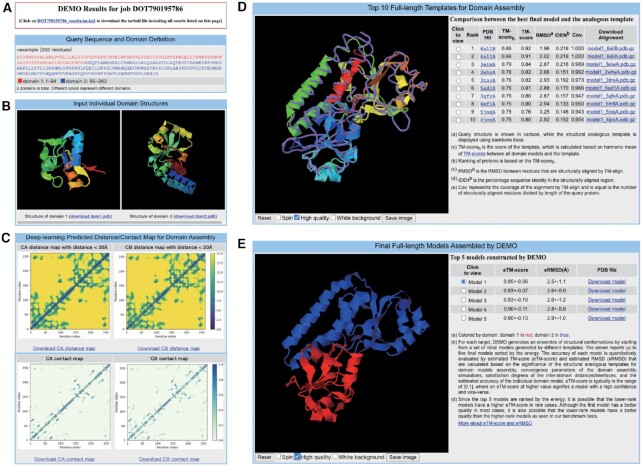
Most proteins in nature contain multiple folding units (or domains). The revolutionary success of AlphaFold2 in single-domain structure prediction showed potential to extend deep-learning techniques for multi-domain structure modeling. This work presents a significantly improved method, DEMO2, which integrates analogous template structural alignments with deep-learning techniques for high-accuracy domain structure assembly. Starting from individual domain models, inter-domain spatial restraints are first predicted with deep residual convolutional networks, where full-length structure models are assembled using L-BFGS simulations under the guidance of a hybrid energy function combining deep-learning restraints and analogous multi-domain template alignments searched from the PDB. The output of DEMO2 contains deep-learning inter-domain restraints, top-ranked multi-domain structure templates, and up to five full-length structure models. DEMO2 was tested on a large-scale benchmark and the blind CASP14 experiment, where DEMO2 was shown to significantly outperform its predecessor and the state-of-the-art protein structure prediction methods. By integrating with new deep-learning techniques, DEMO2 should help fill the rapidly increasing gap between the improved ability of tertiary structure determination and the high demand for the high-quality multi-domain protein structures. The DEMO2 server is available at https://zhanggroup.org/DEMO/.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Recent Progress of Protein Tertiary Structure Prediction.Molecules. 2024 Feb 13;29(4):832. doi: 10.3390/molecules29040832. Molecules. 2024. PMID: 38398585 Free PMC article. Review.
-
LOMETS3: integrating deep learning and profile alignment for advanced protein template recognition and function annotation.Nucleic Acids Res. 2022 Jul 5;50(W1):W454-W464. doi: 10.1093/nar/gkac248. Nucleic Acids Res. 2022. PMID: 35420129 Free PMC article.
-
Protein structure prediction using deep learning distance and hydrogen-bonding restraints in CASP14.Proteins. 2021 Dec;89(12):1734-1751. doi: 10.1002/prot.26193. Epub 2021 Aug 7. Proteins. 2021. PMID: 34331351 Free PMC article.
-
MULTICOM2 open-source protein structure prediction system powered by deep learning and distance prediction.Sci Rep. 2021 Jun 23;11(1):13155. doi: 10.1038/s41598-021-92395-6. Sci Rep. 2021. PMID: 34162922 Free PMC article.
-
I-TASSER-MTD: a deep-learning-based platform for multi-domain protein structure and function prediction.Nat Protoc. 2022 Oct;17(10):2326-2353. doi: 10.1038/s41596-022-00728-0. Epub 2022 Aug 5. Nat Protoc. 2022. PMID: 35931779 Review.
Cited by
-
Structural modelling of human complement FHR1 and two of its synthetic derivatives provides insight into their in-vivo functions.Comput Struct Biotechnol J. 2023 Feb 3;21:1473-1486. doi: 10.1016/j.csbj.2023.02.002. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 36851916 Free PMC article.
-
Narrow funnel-like interaction energy distribution is an indicator of specific protein interaction partner.iScience. 2023 May 20;26(6):106911. doi: 10.1016/j.isci.2023.106911. eCollection 2023 Jun 16. iScience. 2023. PMID: 37305691 Free PMC article.
-
Integrating deep learning, threading alignments, and a multi-MSA strategy for high-quality protein monomer and complex structure prediction in CASP15.Proteins. 2023 Dec;91(12):1684-1703. doi: 10.1002/prot.26585. Epub 2023 Aug 31. Proteins. 2023. PMID: 37650367 Free PMC article.
-
Recent Progress of Protein Tertiary Structure Prediction.Molecules. 2024 Feb 13;29(4):832. doi: 10.3390/molecules29040832. Molecules. 2024. PMID: 38398585 Free PMC article. Review.
-
Deep-learning-based single-domain and multidomain protein structure prediction with D-I-TASSER.Nat Biotechnol. 2025 May 23. doi: 10.1038/s41587-025-02654-4. Online ahead of print. Nat Biotechnol. 2025. PMID: 40410405
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
