

Helicobacter cinaedi is a human-adapted lineage in the Helicobacter cinaedi/canicola/'magdeburgensis' complex
- PMID: 35536747
- PMCID: PMC9465070
- DOI: 10.1099/mgen.0.000830
Helicobacter cinaedi is a human-adapted lineage in the Helicobacter cinaedi/canicola/'magdeburgensis' complex
Abstract
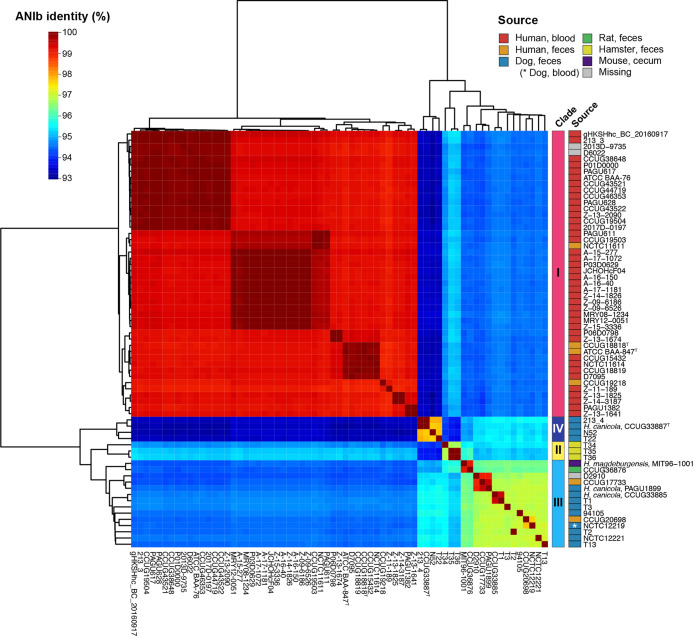
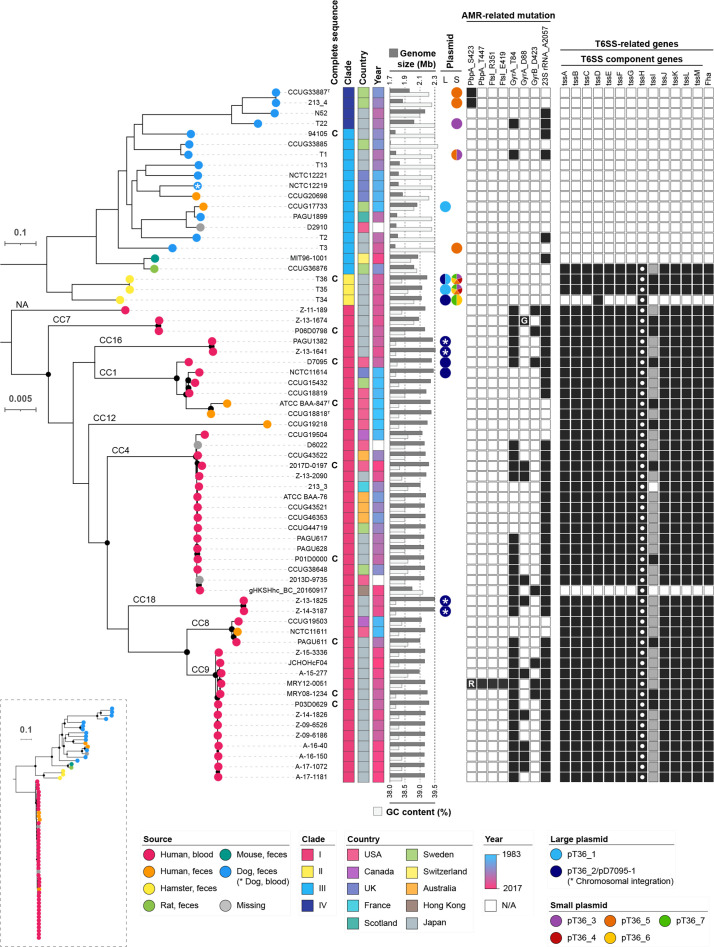
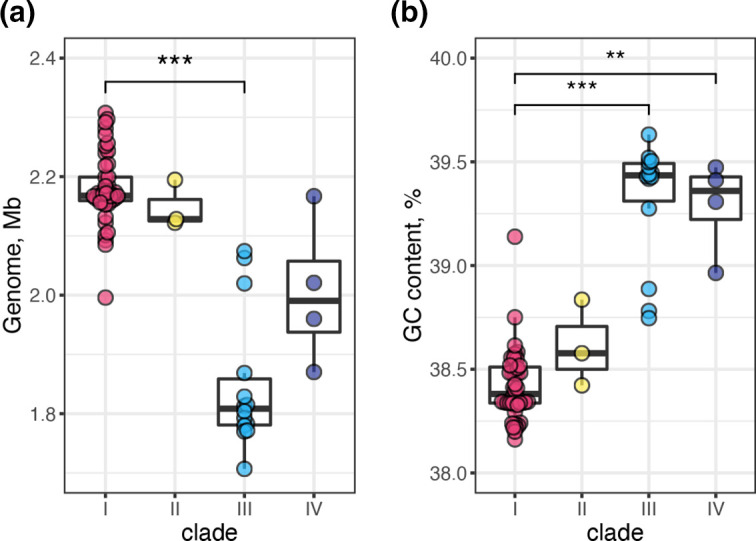
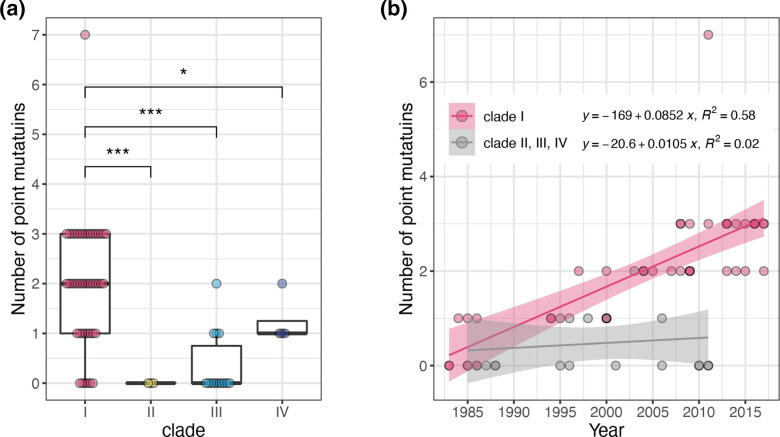
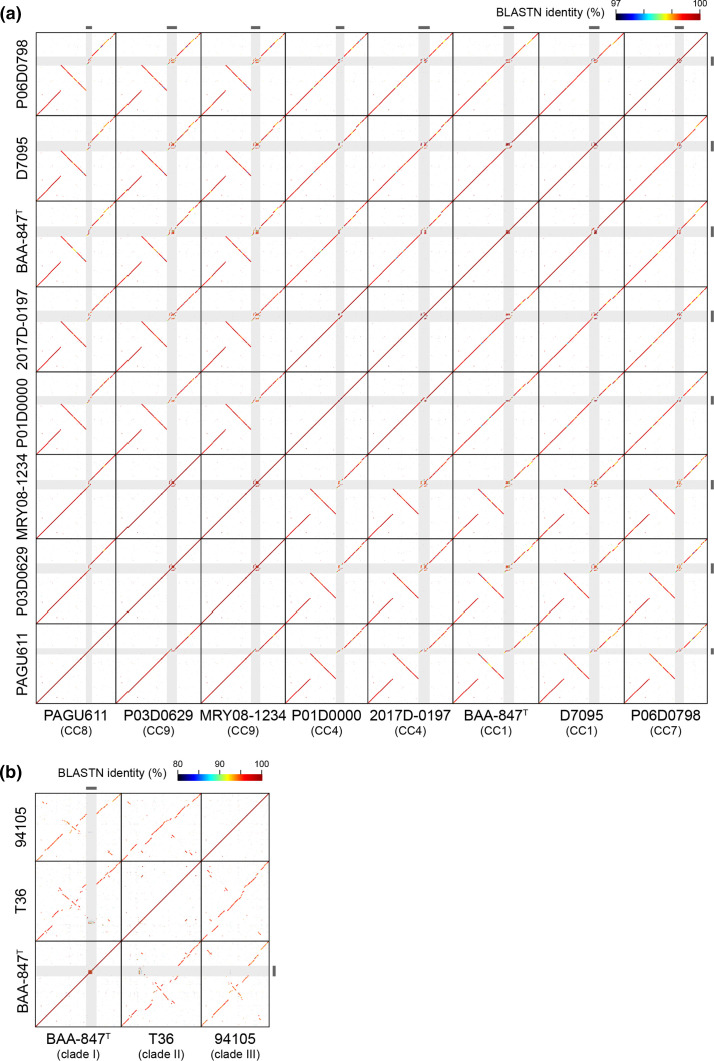
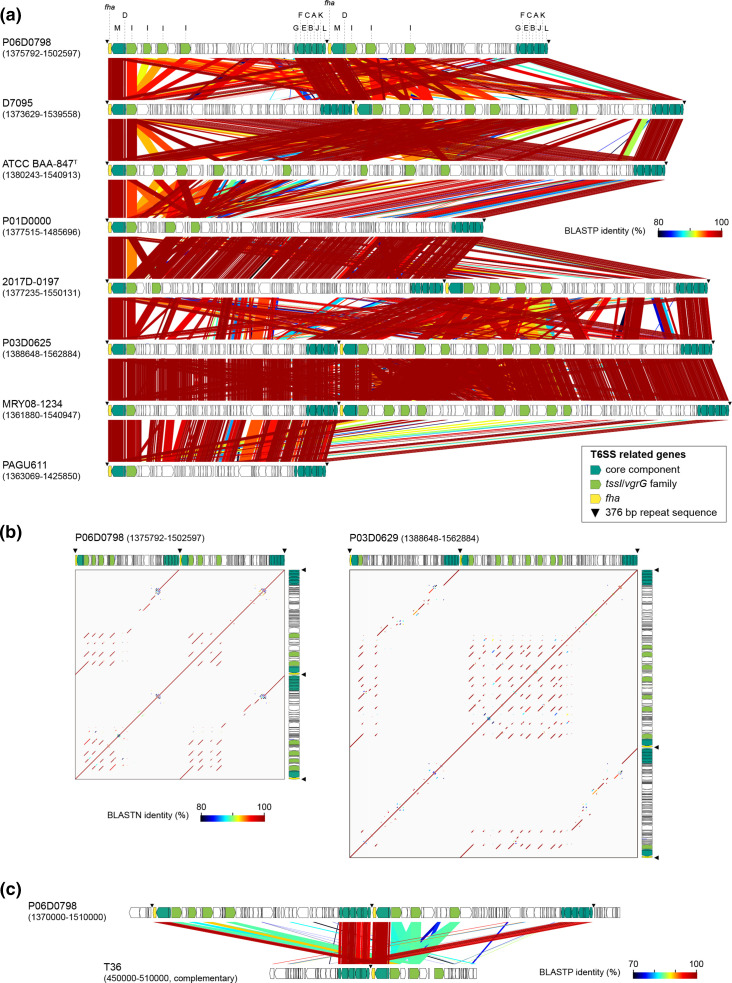
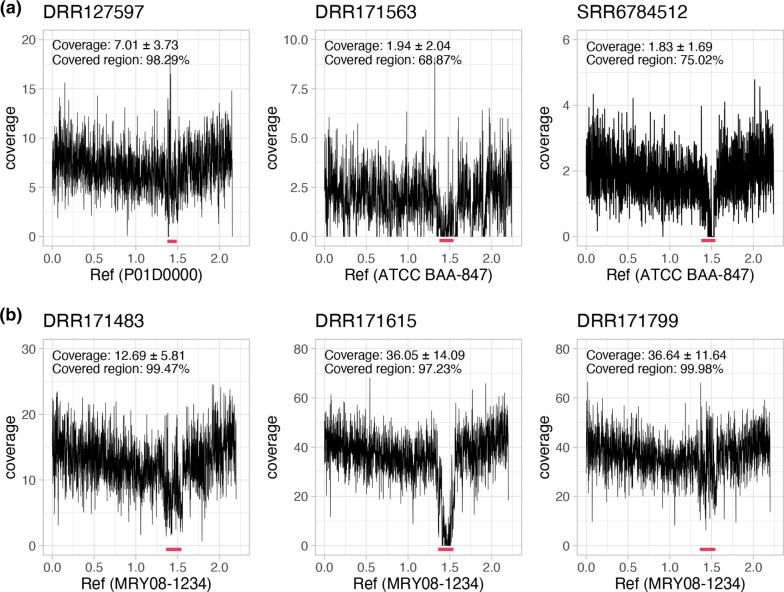
Helicobacter cinaedi is an enterohepatic Helicobacter that causes bacteremia and other diseases in humans. While H. cinaedi-like strains are isolated from animals, including dog isolates belonging to a recently proposed H. canicola, little is known about the genetic differences between H. cinaedi and these animal isolates. Here, we sequenced 43 H. cinaedi- or H. canicola-like strains isolated from humans, hamsters, rats and dogs and collected 81 genome sequences of H. cinaedi, H. canicola and other enterohepatic Helicobacter strains from public databases. Genomic comparison of these strains identified four distinct clades (clades I-IV) in H. cinaedi/canicola/'magderbugensis' (HCCM) complex. Among these, clade I corresponds to H. cinaedi sensu stricto and represents a human-adapted lineage in the complex. We identified several genomic features unique to clade I. They include the accumulation of antimicrobial resistance-related mutations that reflects the human association of clade I and the larger genome size and the presence of a CRISPR-Cas system and multiple toxin-antitoxin and restriction-modification systems, both of which indicate the contribution of horizontal gene transfer to the evolution of clade I. In addition, nearly all clade I strains but only a few strains belonging to one minor clade contained a highly variable genomic region encoding a type VI secretion system (T6SS), which could play important roles in gut colonization by killing competitors or inhibiting their growth. We also developed a method to systematically search for H. cinaedi sequences in large metagenome data sets based on the results of genome comparison. Using this method, we successfully identified multiple HCCM complex-containing human faecal metagenome samples and obtained the sequence information covering almost the entire genome of each strain. Importantly, all were clade I strains, supporting our conclusion that H. cinaedi sensu stricto is a human-adapted lineage in the HCCM complex.
Keywords: Helicobacter canicola; Helicobacter cinaedi; Type VI secretion system; enterohepatic Helicobacter; genome comparison; human adaptation; metagenome data search.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical