

Hypoxia-Reoxygenation Couples 3βHSD1 Enzyme and Cofactor Upregulation to Facilitate Androgen Biosynthesis and Hormone Therapy Resistance in Prostate Cancer
- PMID: 35536859
- PMCID: PMC9256813
- DOI: 10.1158/0008-5472.CAN-21-4256
Hypoxia-Reoxygenation Couples 3βHSD1 Enzyme and Cofactor Upregulation to Facilitate Androgen Biosynthesis and Hormone Therapy Resistance in Prostate Cancer
Abstract
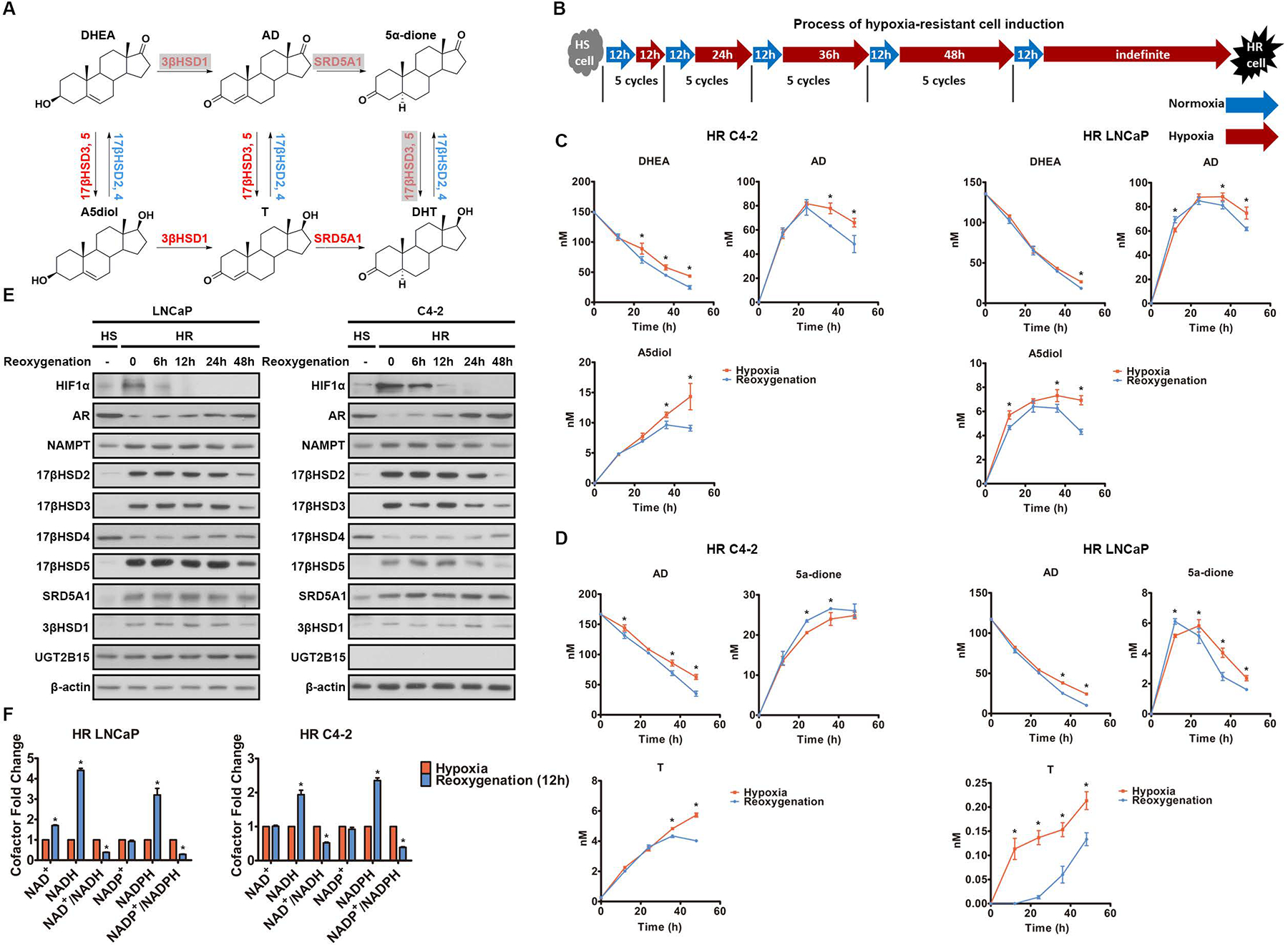
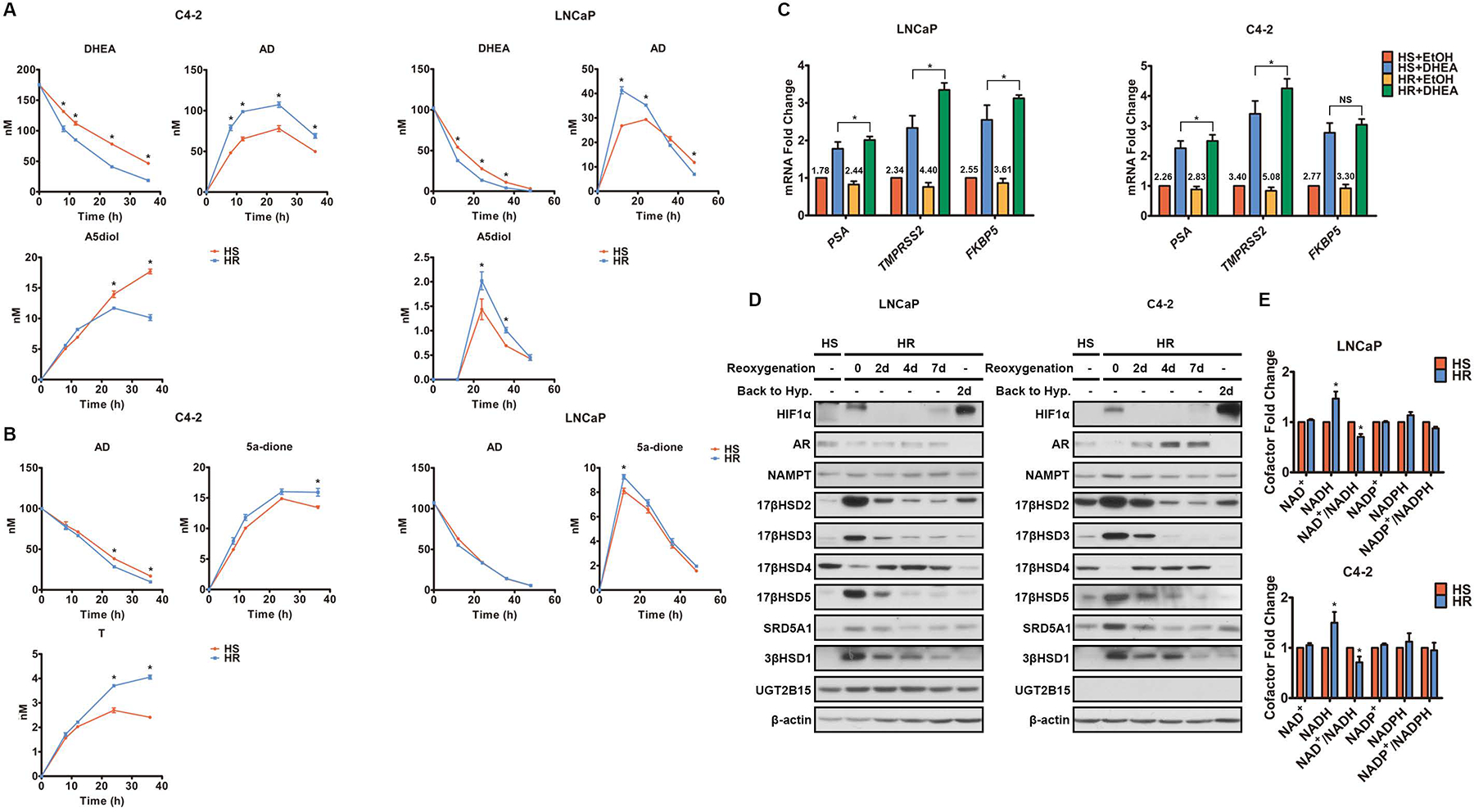
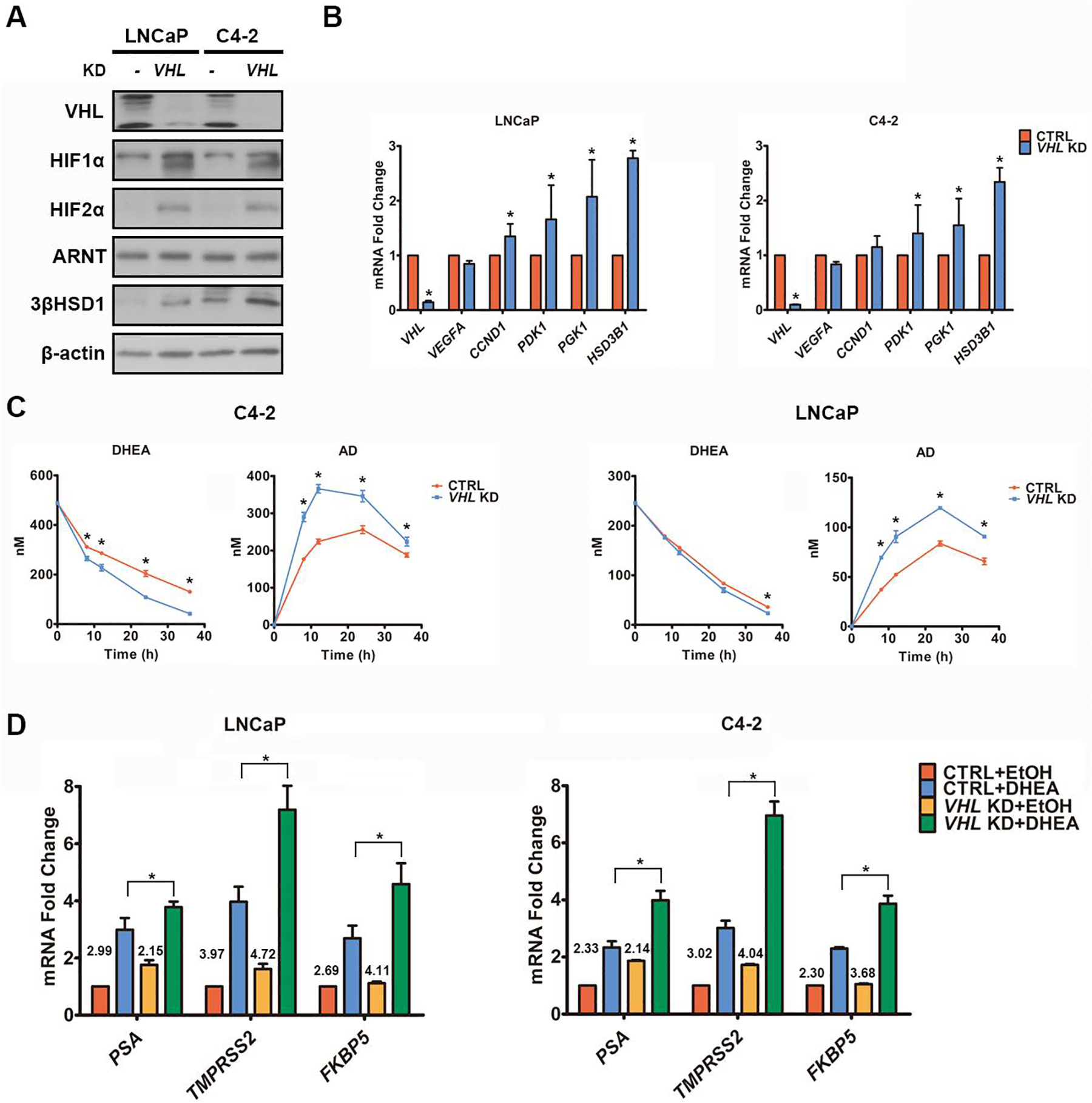
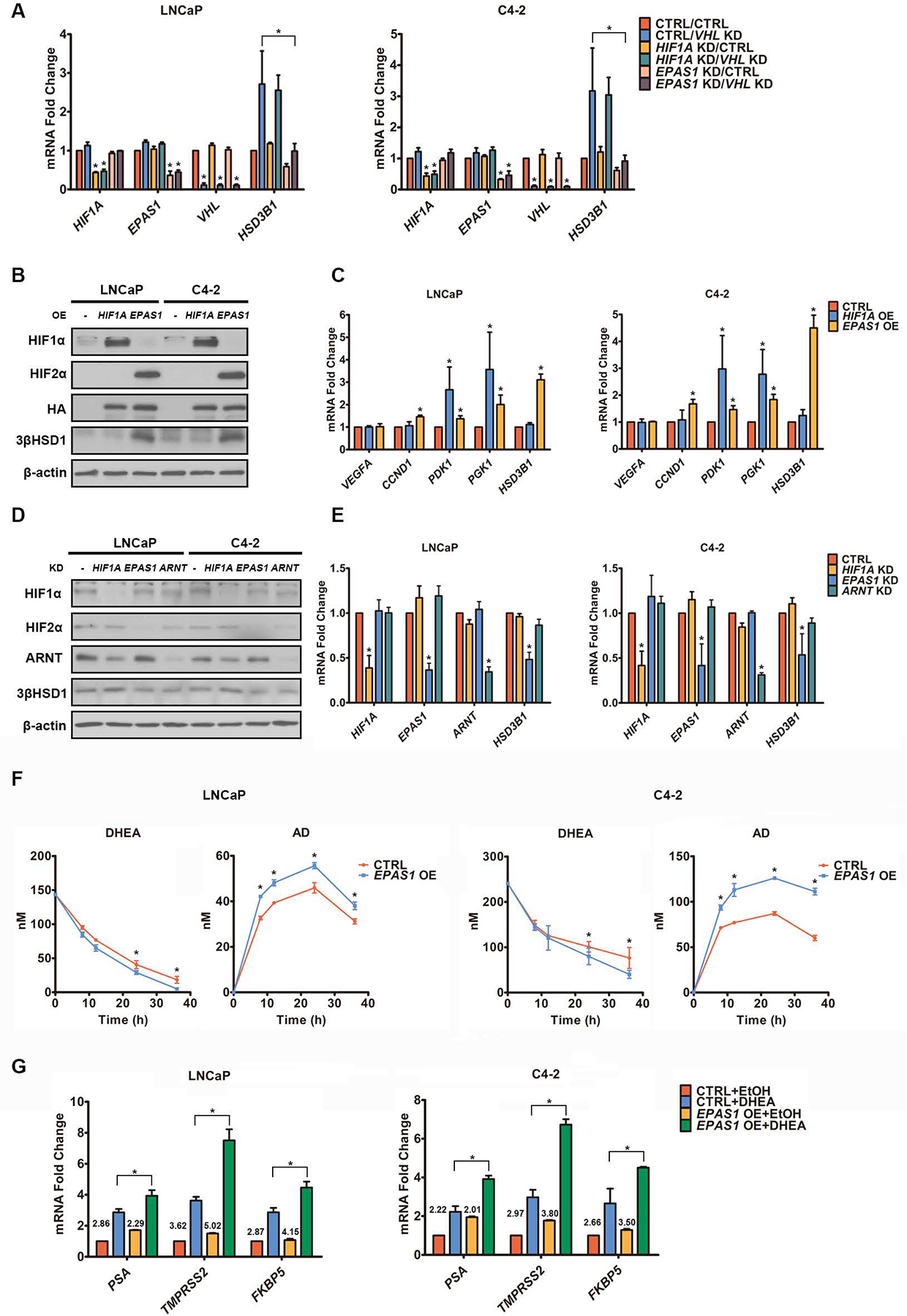
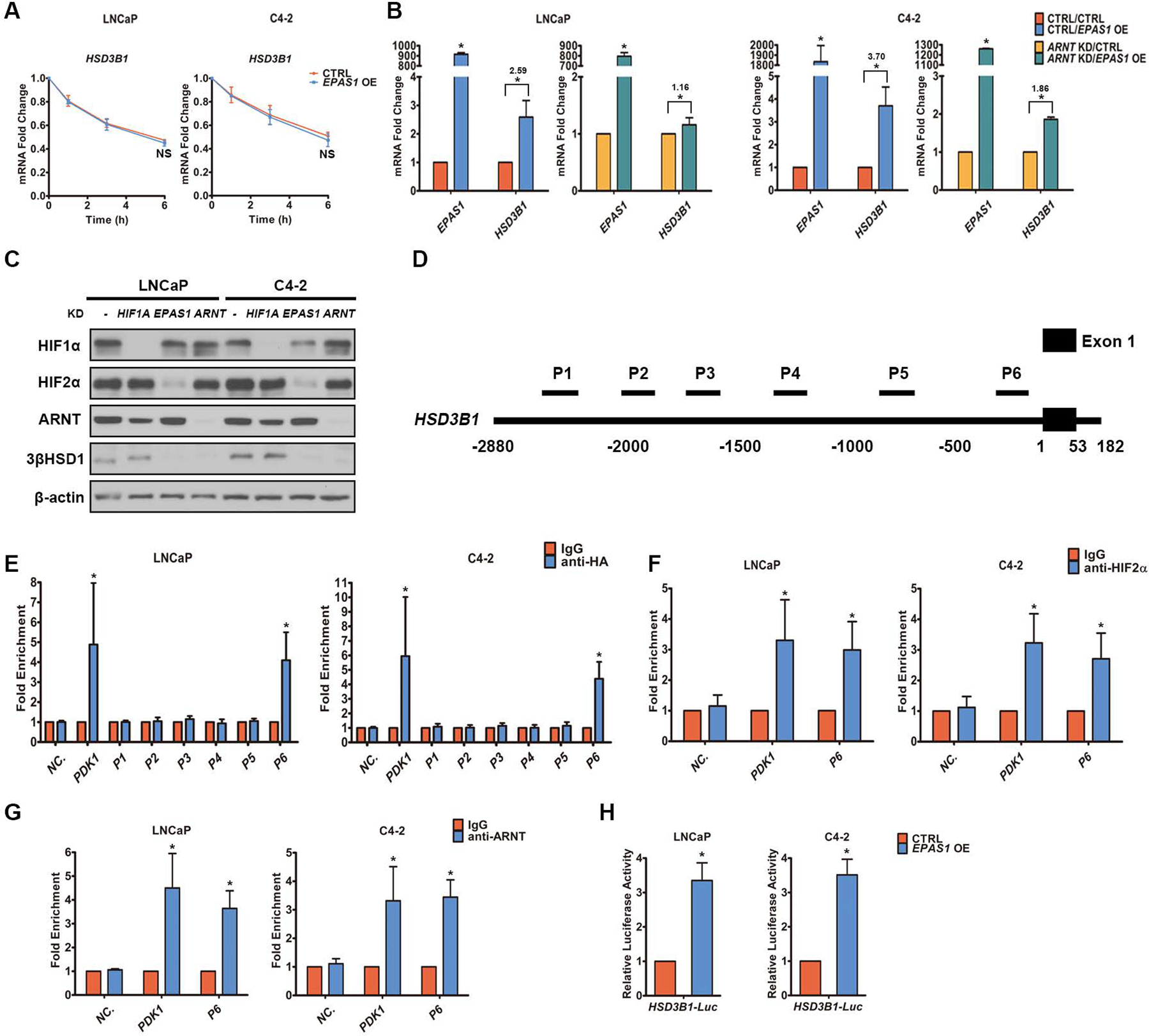
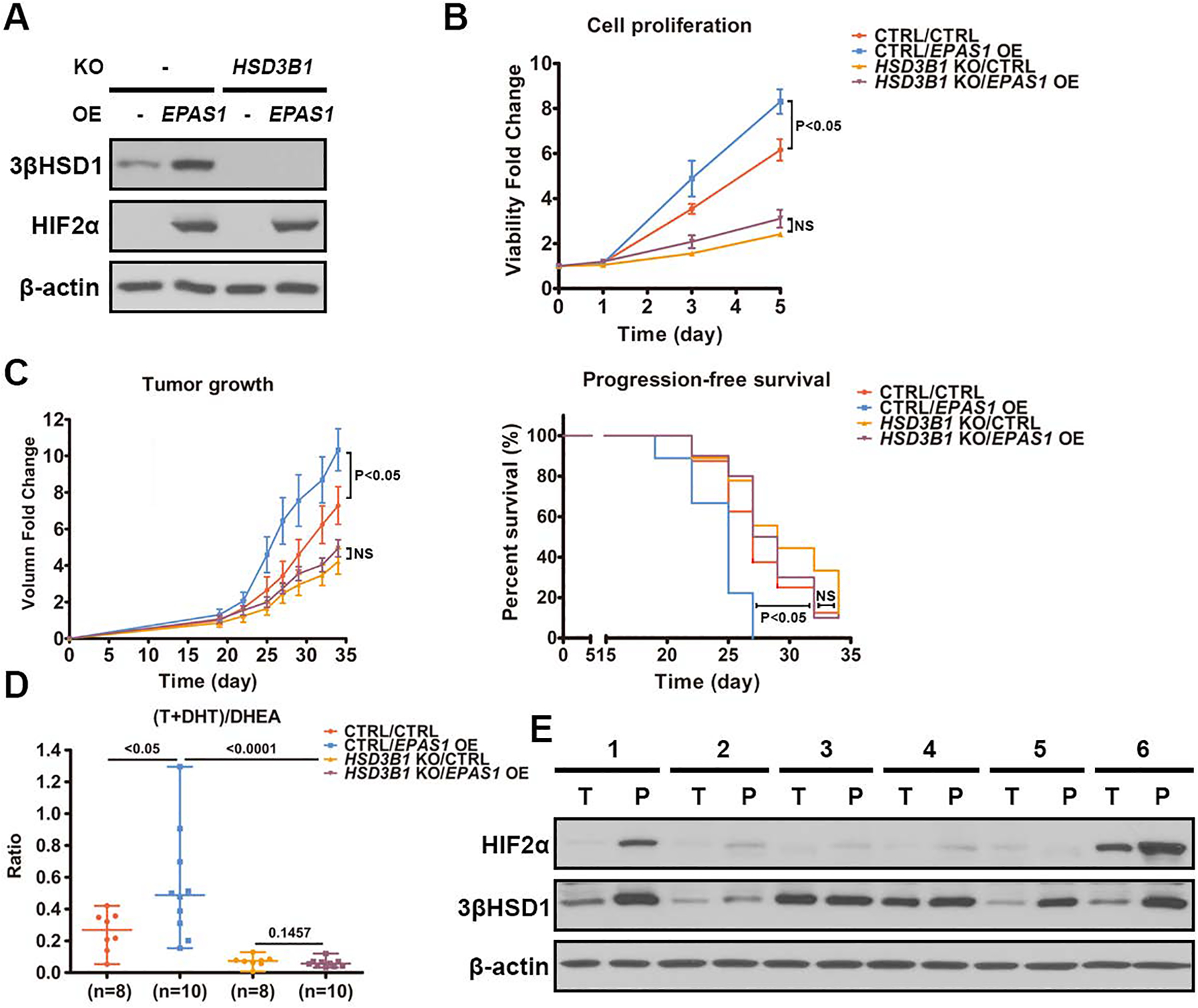
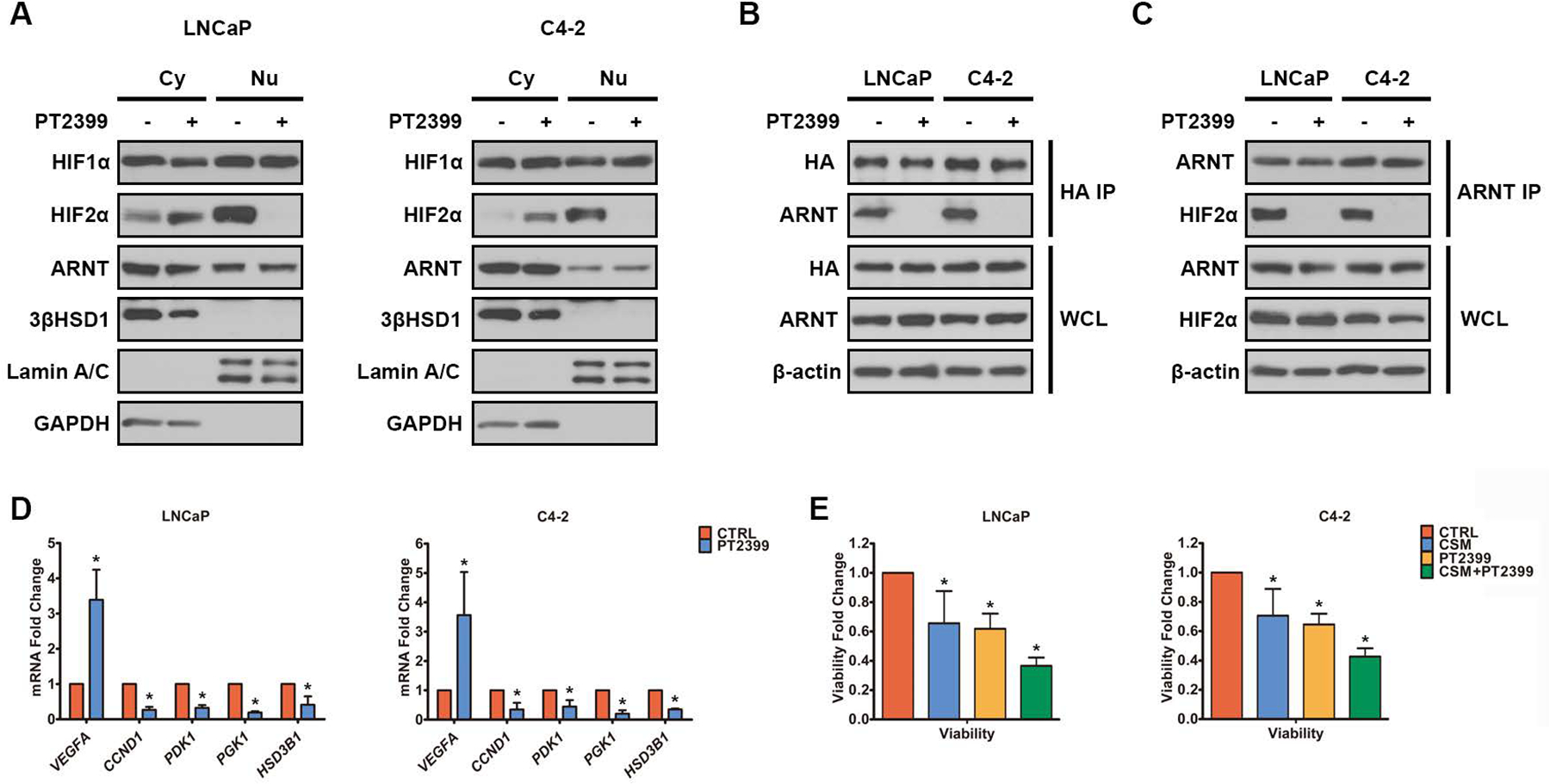
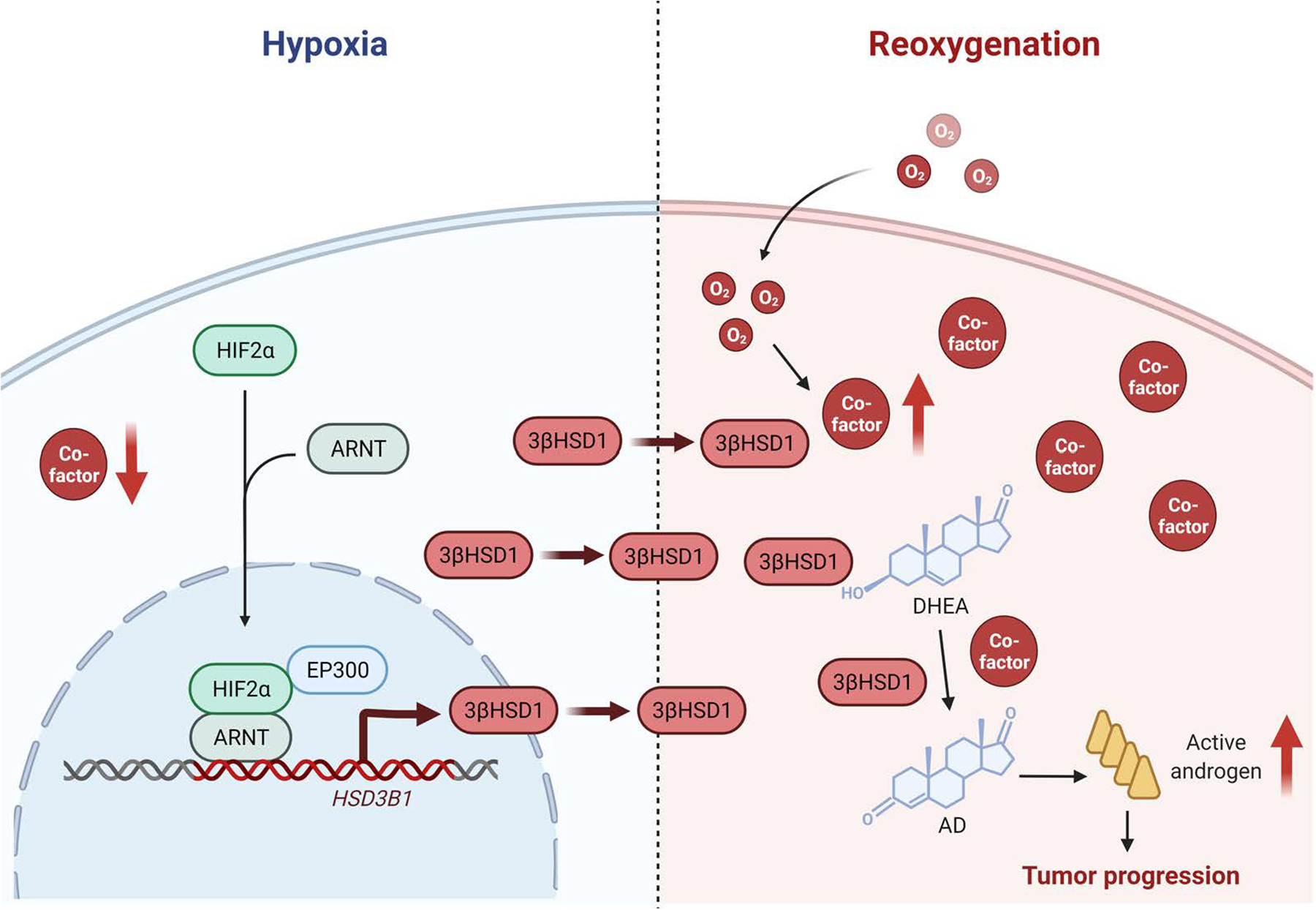
Androgen deprivation therapy suppresses tumor androgen receptor (AR) signaling by depleting circulating testosterone and is a mainstay treatment for advanced prostate cancer. Despite initial treatment response, castration-resistant prostate cancer nearly always develops and remains driven primarily by the androgen axis. Here we investigated how changes in oxygenation affect androgen synthesis. In prostate cancer cells, chronic hypoxia coupled to reoxygenation resulted in efficient metabolism of androgen precursors to produce androgens and activate AR. Hypoxia induced 3βHSD1, the rate-limiting androgen synthesis regulator, and reoxygenation replenished necessary cofactors, suggesting that hypoxia and reoxygenation both facilitate potent androgen synthesis. The EGLN1/VHL/HIF2α pathway induced 3βHSD1 expression through direct binding of HIF2α to the 5' regulatory region of HSD3B1 to promote transcription. Overexpression of HIF2α facilitated prostate cancer progression, which largely depended on 3βHSD1. Inhibition of HIF2α with the small-molecule PT2399 prevented prostate cancer cell proliferation. These results thus identify HIF2α as a regulator of androgen synthesis and potential therapeutic target in prostate cancer.
Significance: Hypoxia followed by reoxygenation in prostate cancer drives androgen deprivation therapy resistance via increasing the rate-limiting enzyme and cofactors for androgen synthesis, revealing HIF2α as a therapeutic target to subvert resistance.
©2022 American Association for Cancer Research.
Conflict of interest statement
Conflict of interest
None of the authors have a conflict of interest associated with the work presented here.
Figures
Comment in
-
Uro-Science.J Urol. 2023 Apr;209(4):801-802. doi: 10.1097/JU.0000000000003161. Epub 2023 Jan 19. J Urol. 2023. PMID: 36655472 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
