

1H-Detected Biomolecular NMR under Fast Magic-Angle Spinning
- PMID: 35536915
- PMCID: PMC9136936
- DOI: 10.1021/acs.chemrev.1c00918
1H-Detected Biomolecular NMR under Fast Magic-Angle Spinning
Abstract
Since the first pioneering studies on small deuterated peptides dating more than 20 years ago, 1H detection has evolved into the most efficient approach for investigation of biomolecular structure, dynamics, and interactions by solid-state NMR. The development of faster and faster magic-angle spinning (MAS) rates (up to 150 kHz today) at ultrahigh magnetic fields has triggered a real revolution in the field. This new spinning regime reduces the 1H-1H dipolar couplings, so that a direct detection of 1H signals, for long impossible without proton dilution, has become possible at high resolution. The switch from the traditional MAS NMR approaches with 13C and 15N detection to 1H boosts the signal by more than an order of magnitude, accelerating the site-specific analysis and opening the way to more complex immobilized biological systems of higher molecular weight and available in limited amounts. This paper reviews the concepts underlying this recent leap forward in sensitivity and resolution, presents a detailed description of the experimental aspects of acquisition of multidimensional correlation spectra with fast MAS, and summarizes the most successful strategies for the assignment of the resonances and for the elucidation of protein structure and conformational dynamics. It finally outlines the many examples where 1H-detected MAS NMR has contributed to the detailed characterization of a variety of crystalline and noncrystalline biomolecular targets involved in biological processes ranging from catalysis through drug binding, viral infectivity, amyloid fibril formation, to transport across lipid membranes.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
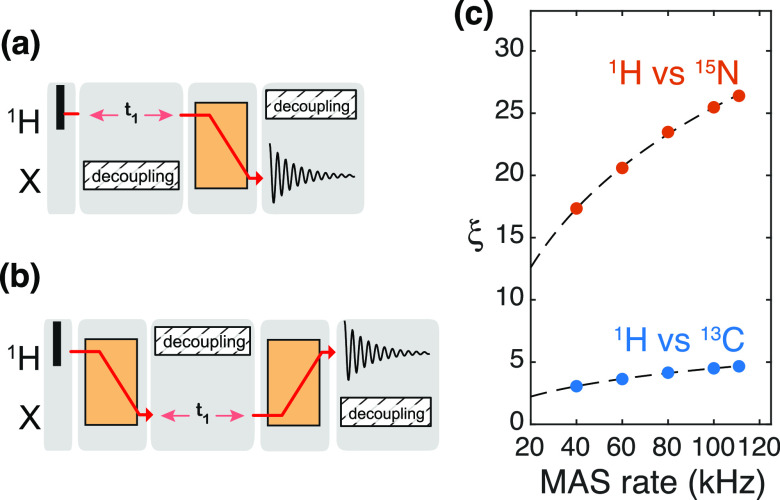
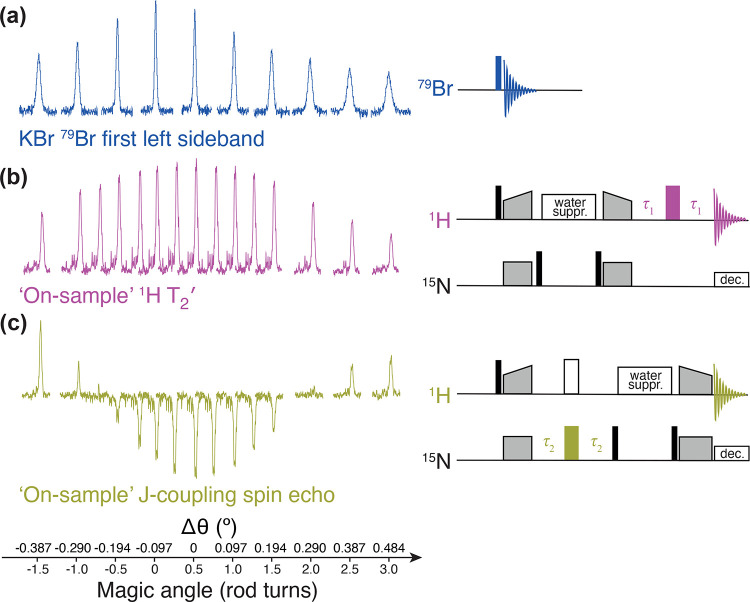
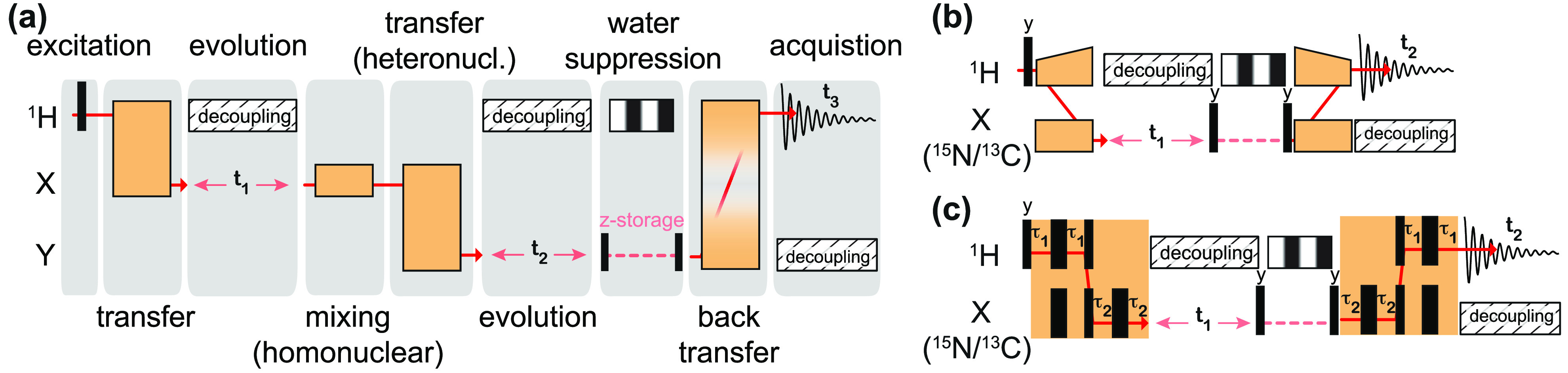
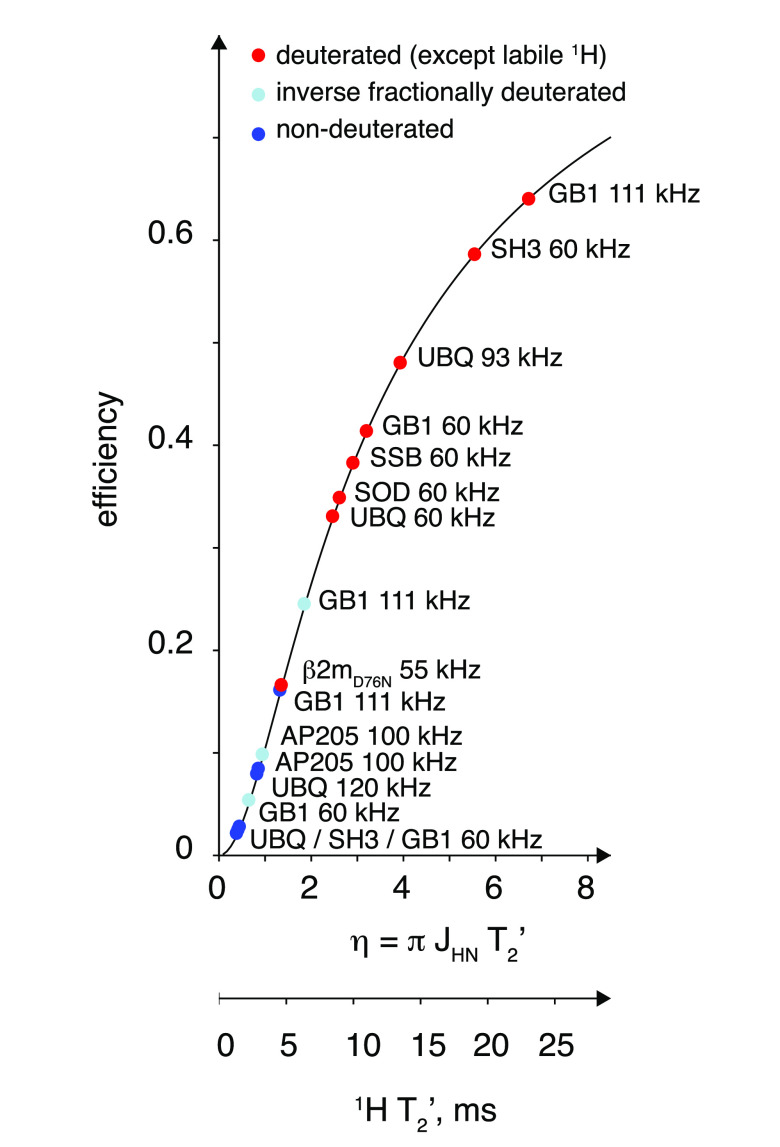
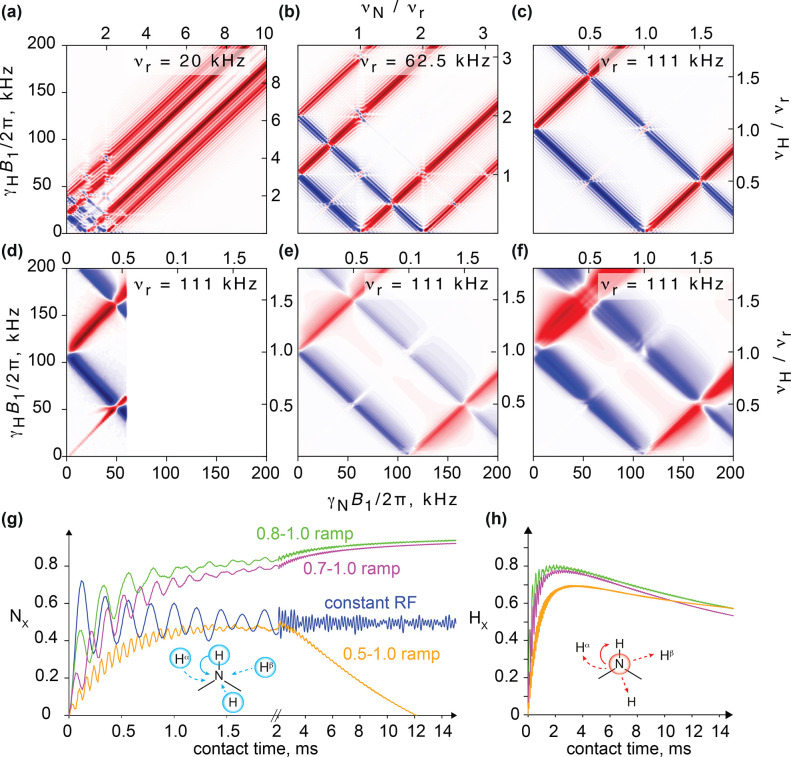
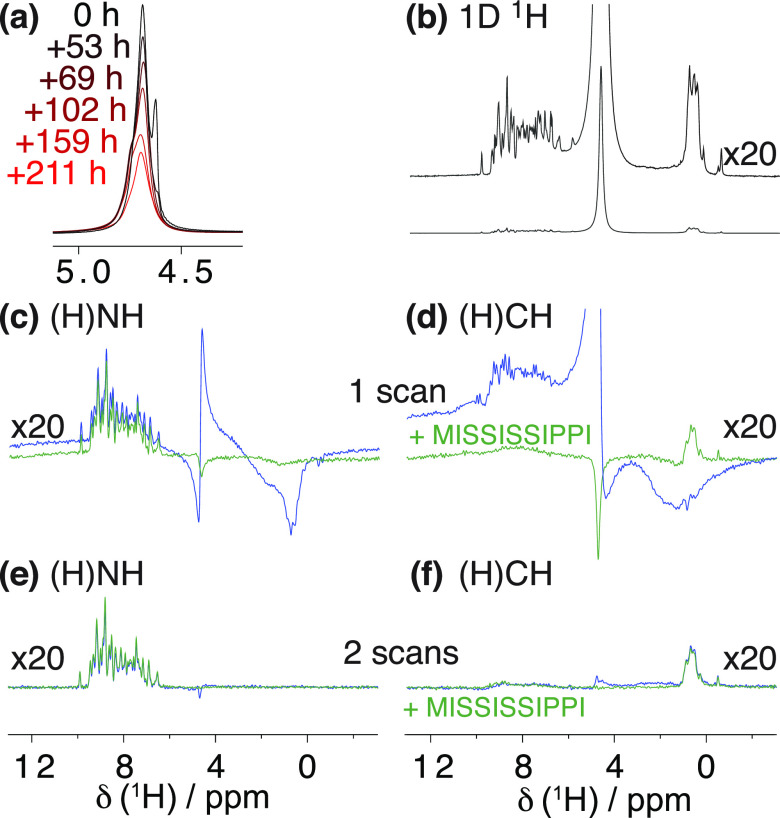
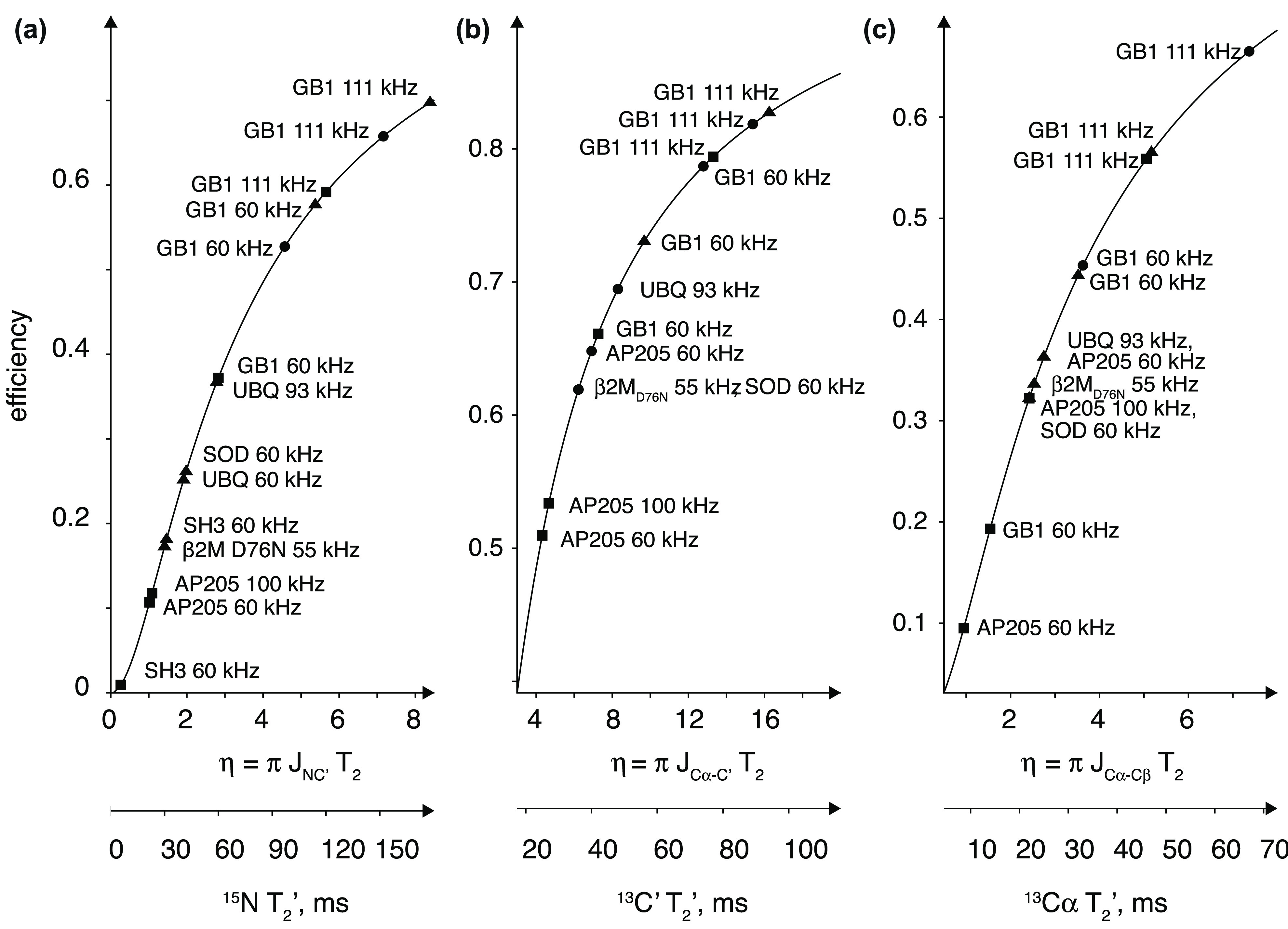
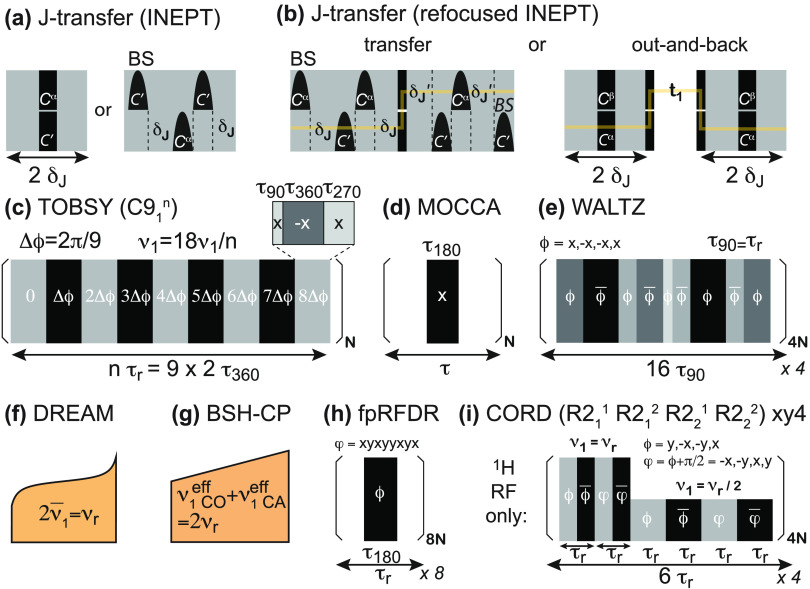
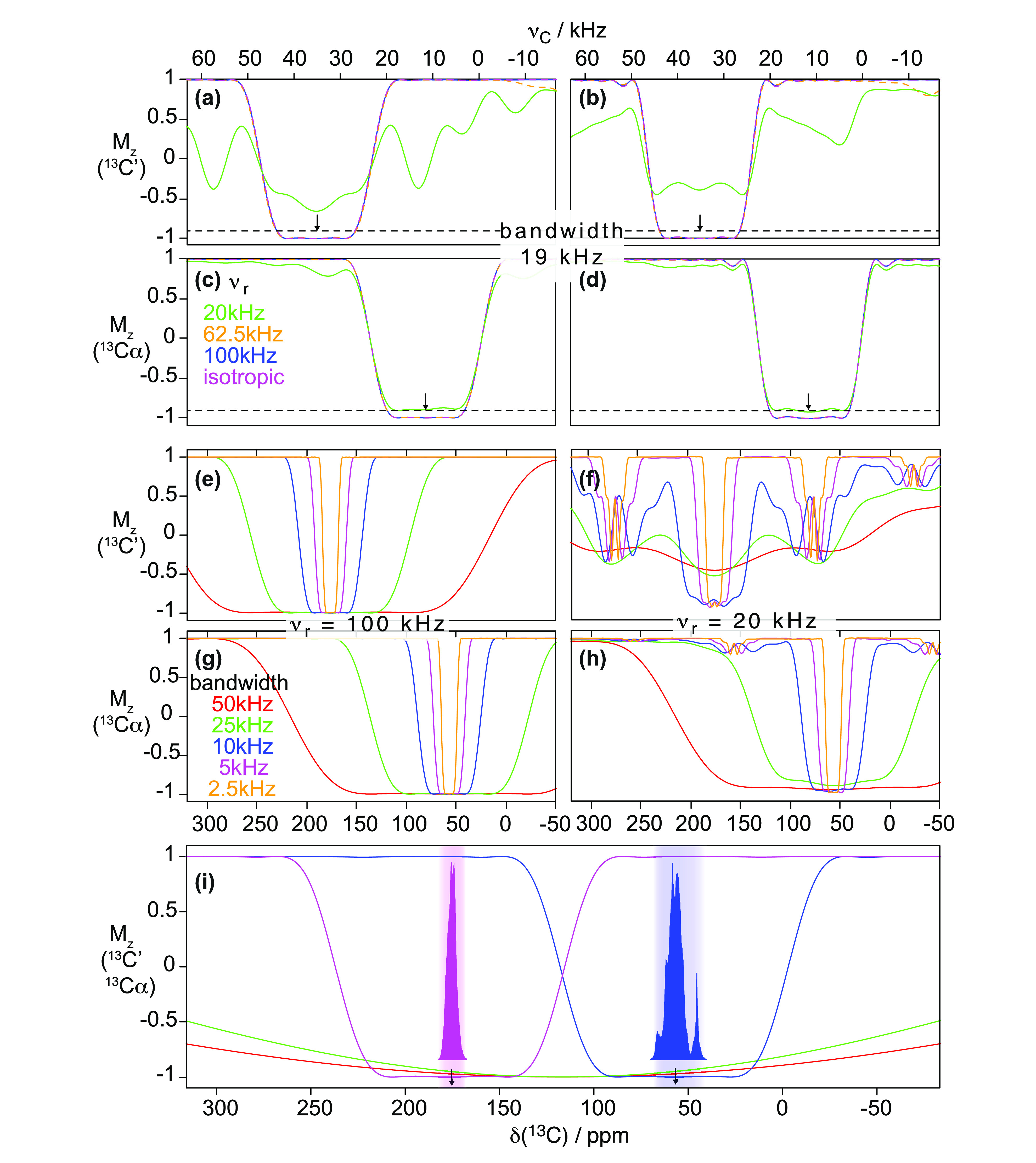
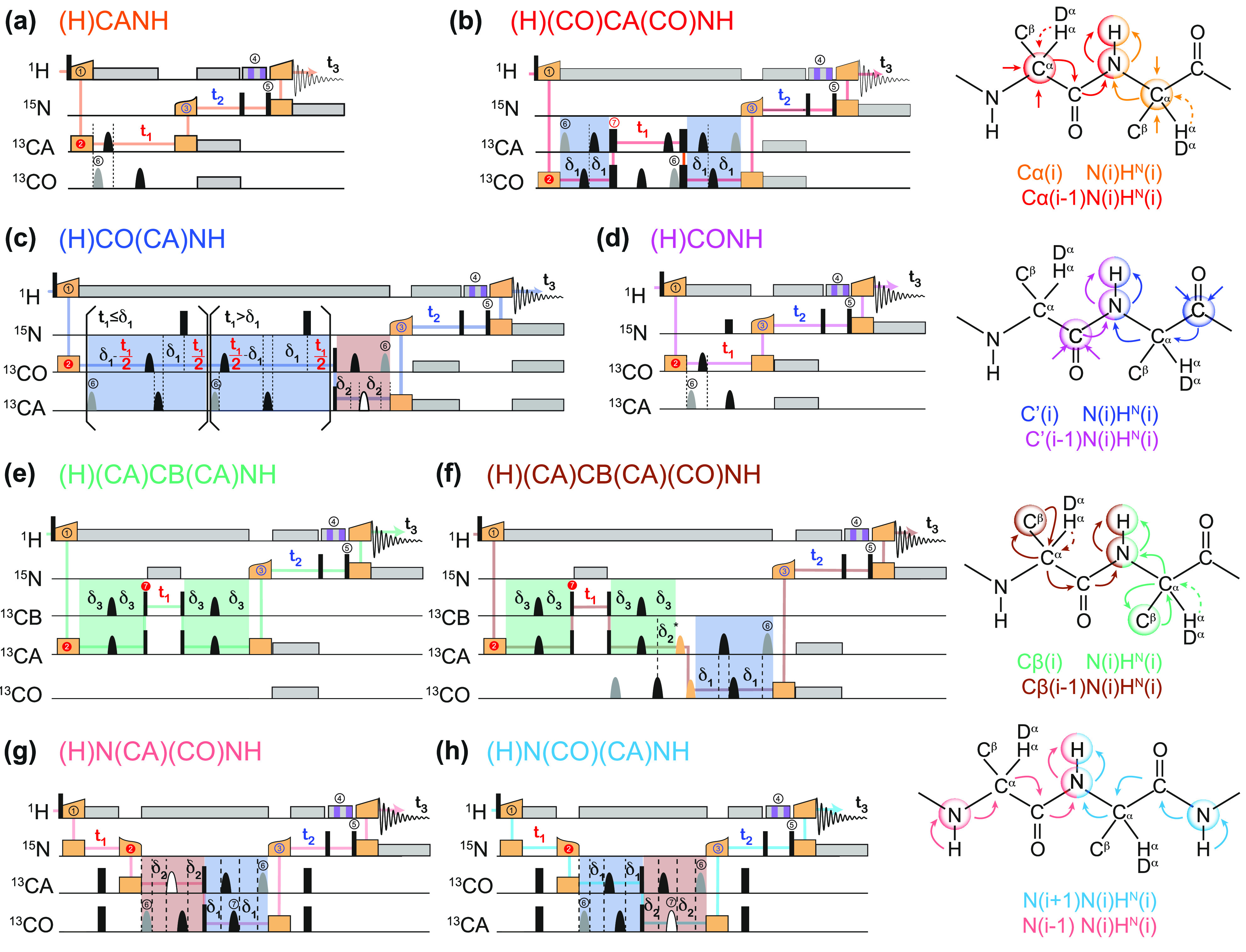

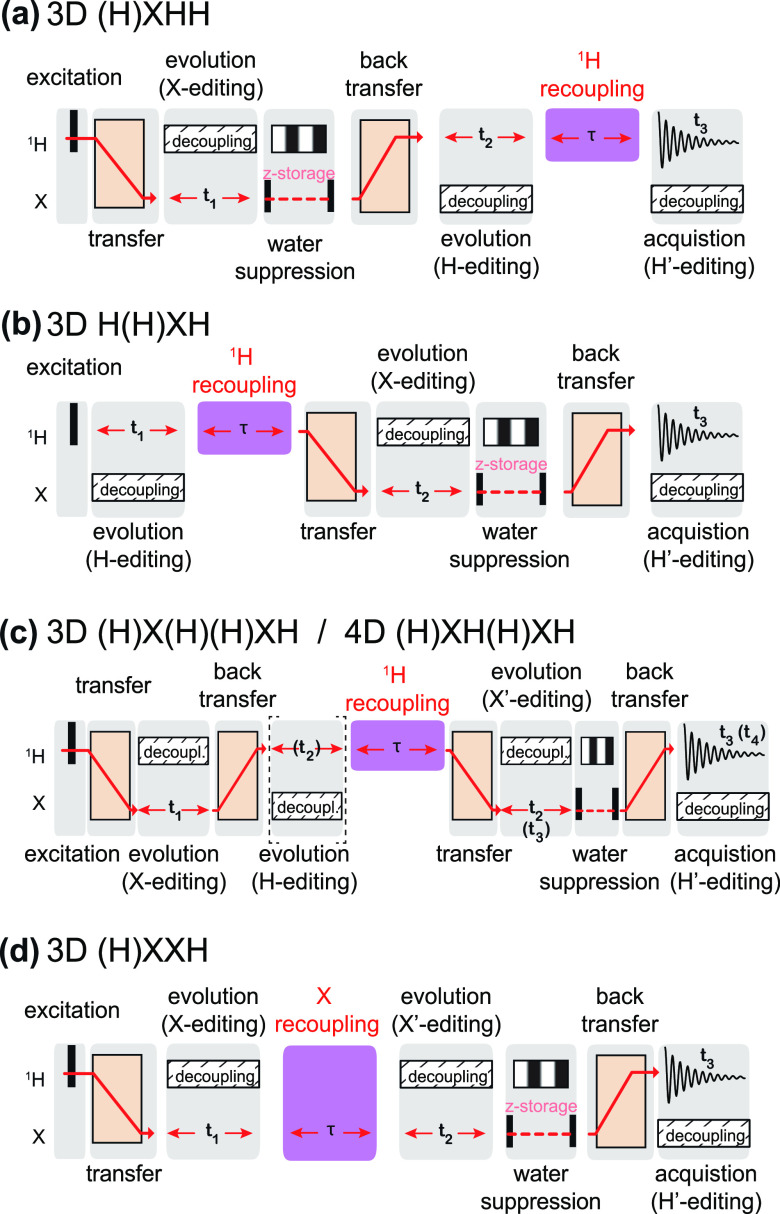
 , ②
=
, ②
=  , ③
=
, ③
=  , ⑤ = y. In (b,e,f), ⑦
= x,
and in (g,h), ⑦ = 8(x)8(y). Additionally, in (a−d,f,g,h),
⑥ = xy for suppression of undesired pathways. In (a−f),
ϕrec =
, ⑤ = y. In (b,e,f), ⑦
= x,
and in (g,h), ⑦ = 8(x)8(y). Additionally, in (a−d,f,g,h),
⑥ = xy for suppression of undesired pathways. In (a−f),
ϕrec = 
 , and in (g,h),
ϕrec =
, and in (g,h),
ϕrec = 


 . The full
phase-cycle length of 8 for (a−f),
and 16 (g,h) can be reduced to 4 and 8, respectively, with a minimal
loss of quality. The phases of the MISSISSIPPI pulses ④ = xy
. The full
phase-cycle length of 8 for (a−f),
and 16 (g,h) can be reduced to 4 and 8, respectively, with a minimal
loss of quality. The phases of the MISSISSIPPI pulses ④ = xy are not cycled. Open and filled red (or
blue) circles indicate phases which are respectively incremented or
decremented for quadrature detection in t1 (or t2) according to the States-TPPI
procedure. Off-resonance CP or selective pulses are performed with
a fine stepwise phase modulation. The former require appropriate phase
alignment either at the beginning or end of the CP pulse. In (b) and
(c), 13C chemical shifts are evolved off-resonance; however,
folding (aliasing) of resonance frequencies is avoided by t1 time-proportional phase incrementation: Δφ
= 360°ΔΩt1 (in degrees),
ΔΩ = ΩC′/Cα – Δcarrier, where ΩC′/Cα are effective
(requested) centers of either 13C′ or 13Cα bands. In (c), transfer time δ1 is exploited
for a constant-time evolution of 13C′ if t1 < δ1; otherwise, a real-time
mode is used. Panels (a–f) were adapted with permission from
ref (198) (copyright
2014 American Chemical Society) and panels (g−h) from ref (200) (copyright 2015 Springer
Nature).
are not cycled. Open and filled red (or
blue) circles indicate phases which are respectively incremented or
decremented for quadrature detection in t1 (or t2) according to the States-TPPI
procedure. Off-resonance CP or selective pulses are performed with
a fine stepwise phase modulation. The former require appropriate phase
alignment either at the beginning or end of the CP pulse. In (b) and
(c), 13C chemical shifts are evolved off-resonance; however,
folding (aliasing) of resonance frequencies is avoided by t1 time-proportional phase incrementation: Δφ
= 360°ΔΩt1 (in degrees),
ΔΩ = ΩC′/Cα – Δcarrier, where ΩC′/Cα are effective
(requested) centers of either 13C′ or 13Cα bands. In (c), transfer time δ1 is exploited
for a constant-time evolution of 13C′ if t1 < δ1; otherwise, a real-time
mode is used. Panels (a–f) were adapted with permission from
ref (198) (copyright
2014 American Chemical Society) and panels (g−h) from ref (200) (copyright 2015 Springer
Nature).
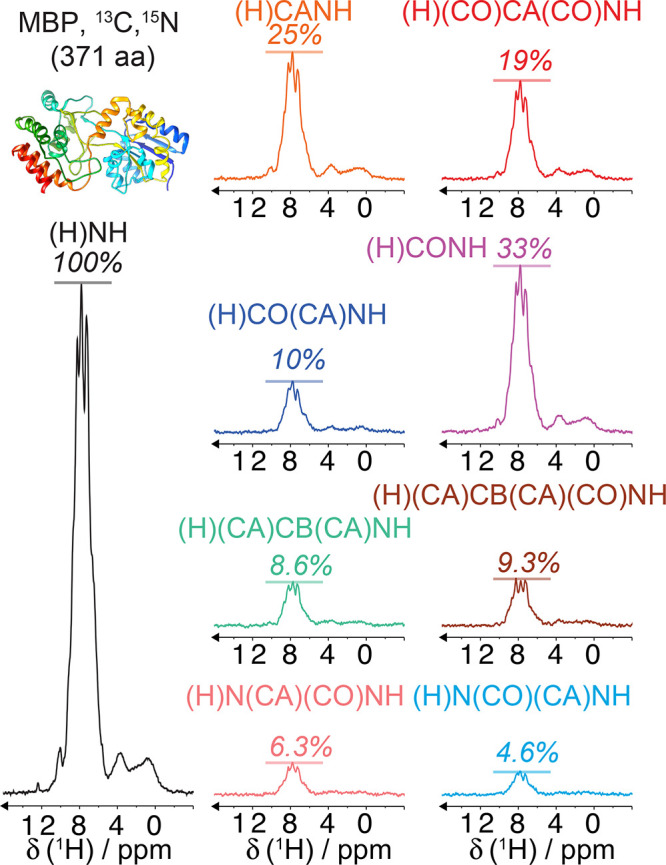
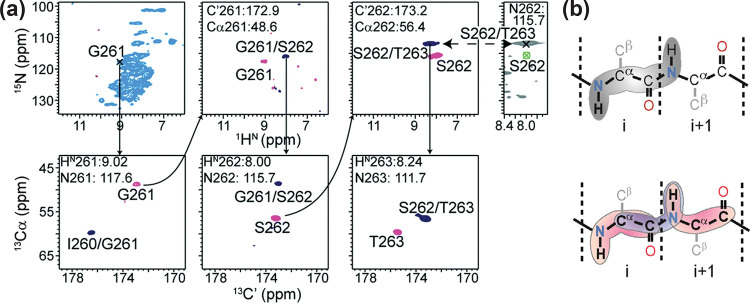
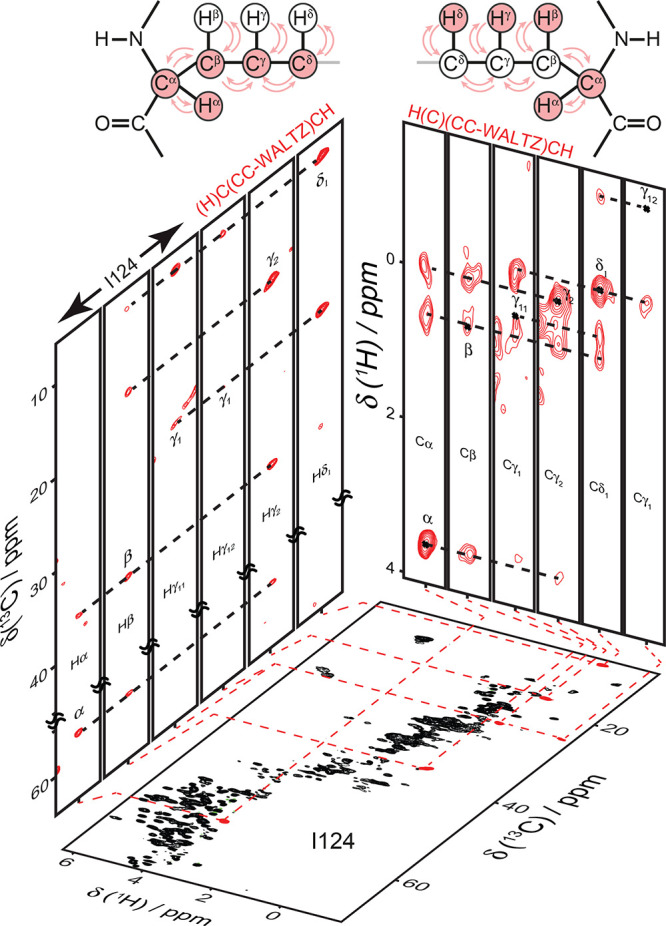
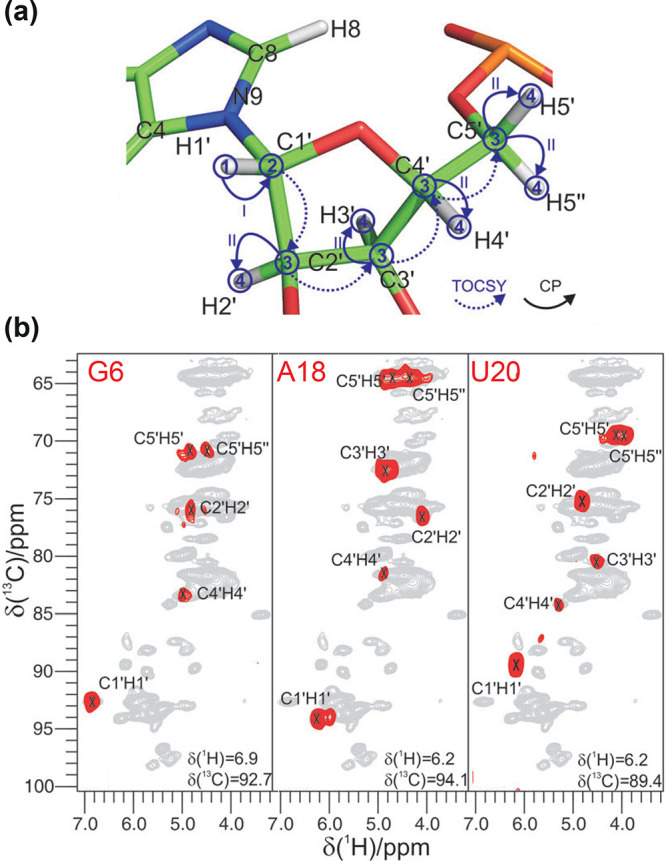
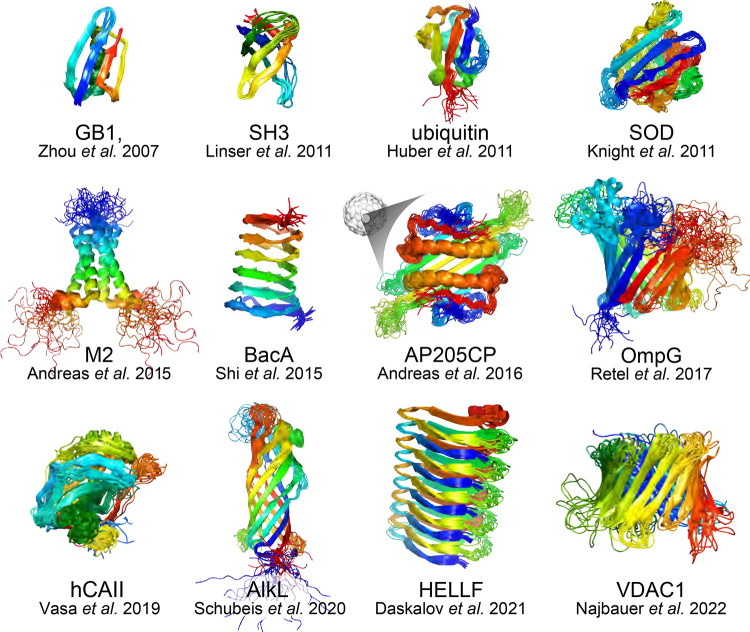
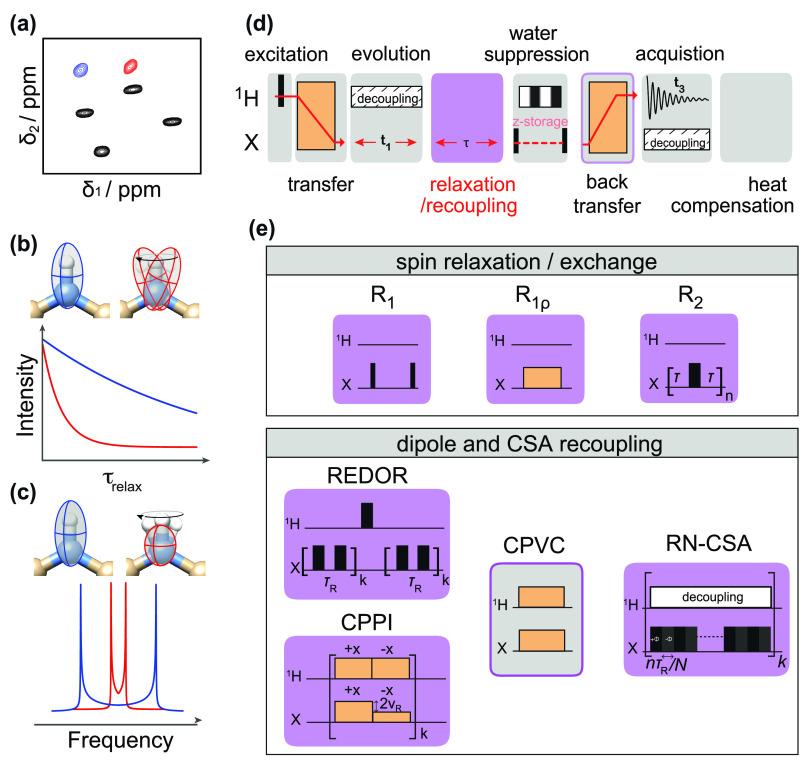
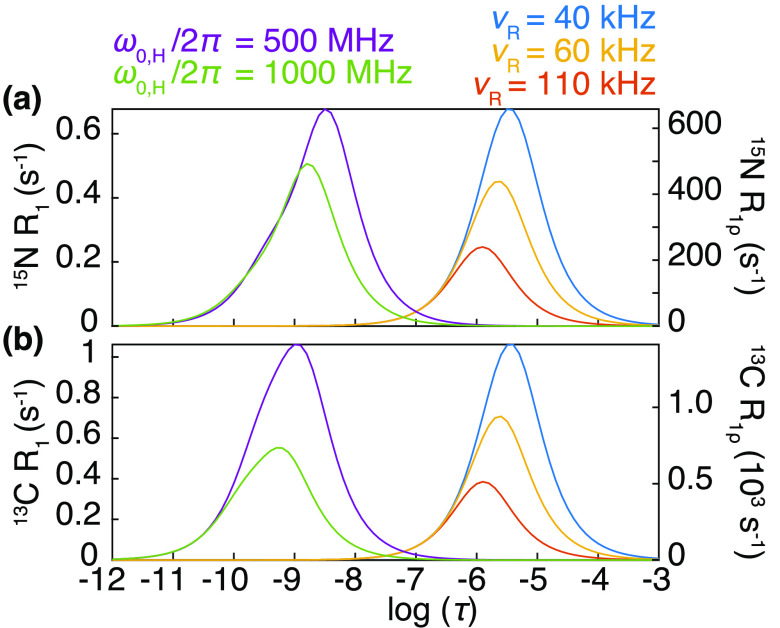
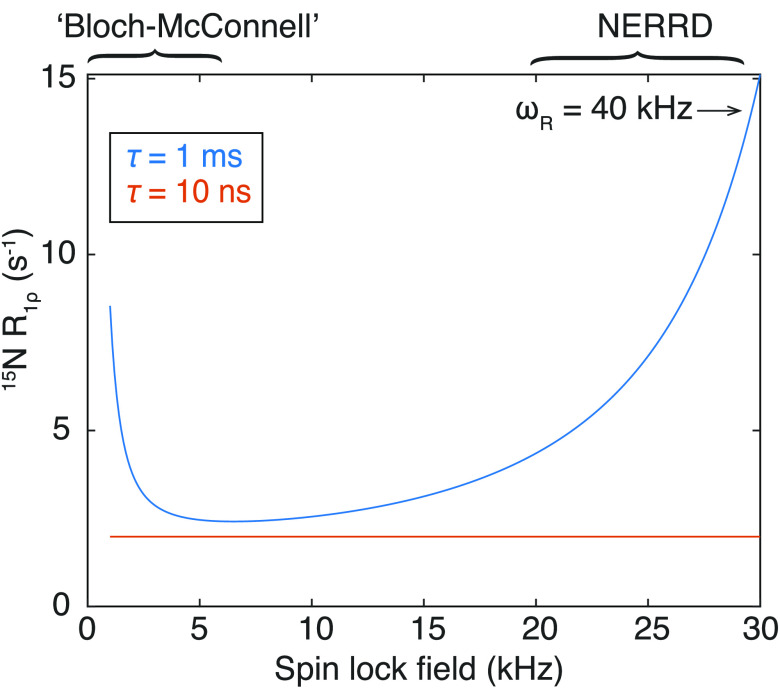
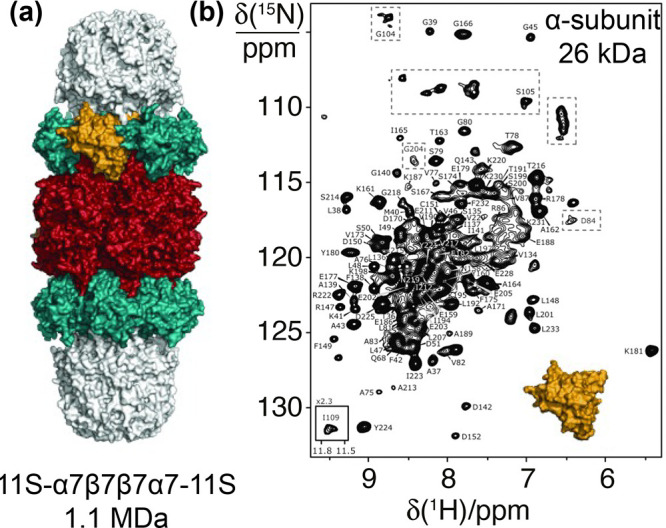
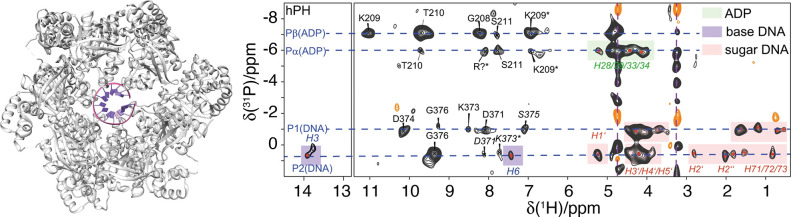
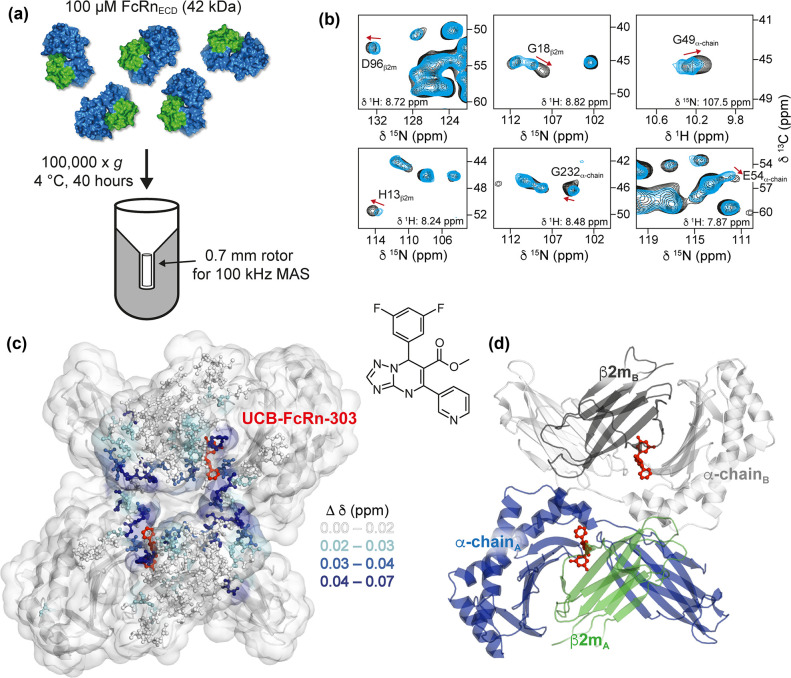
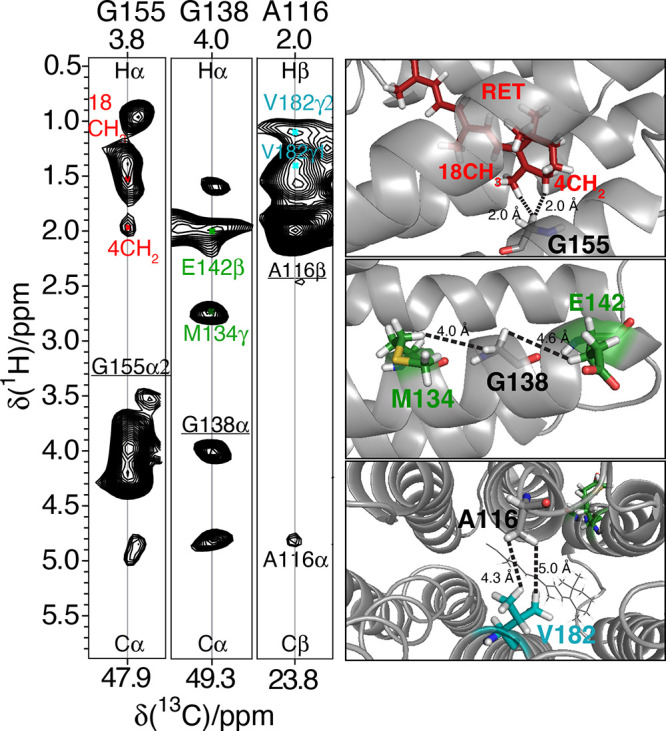
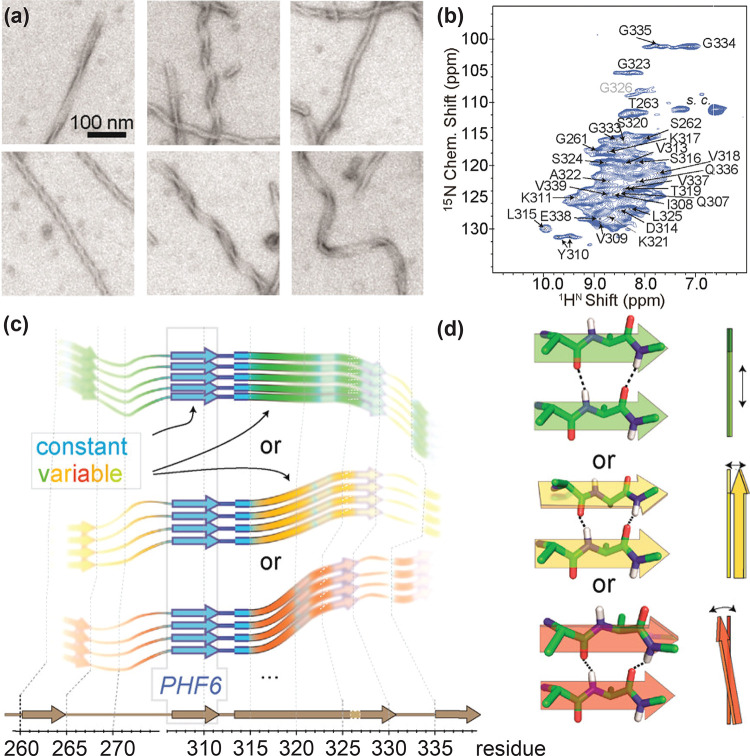
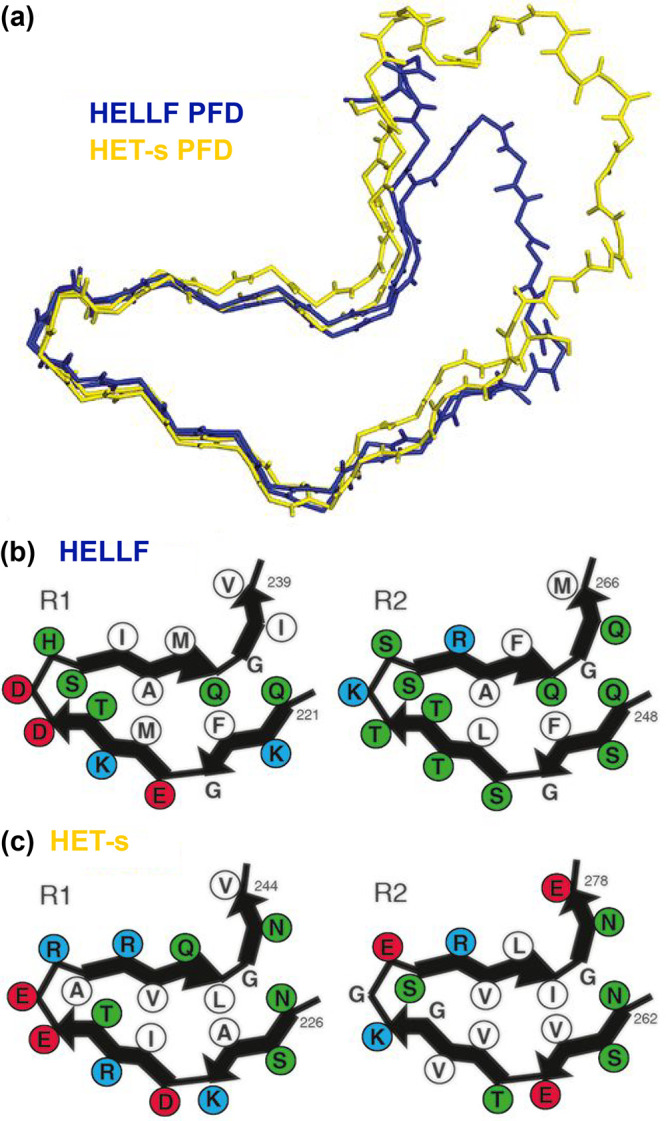
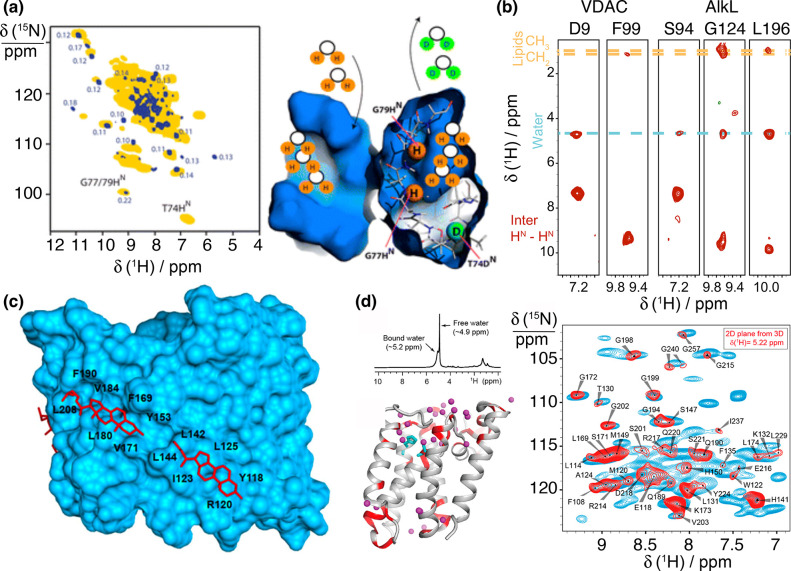
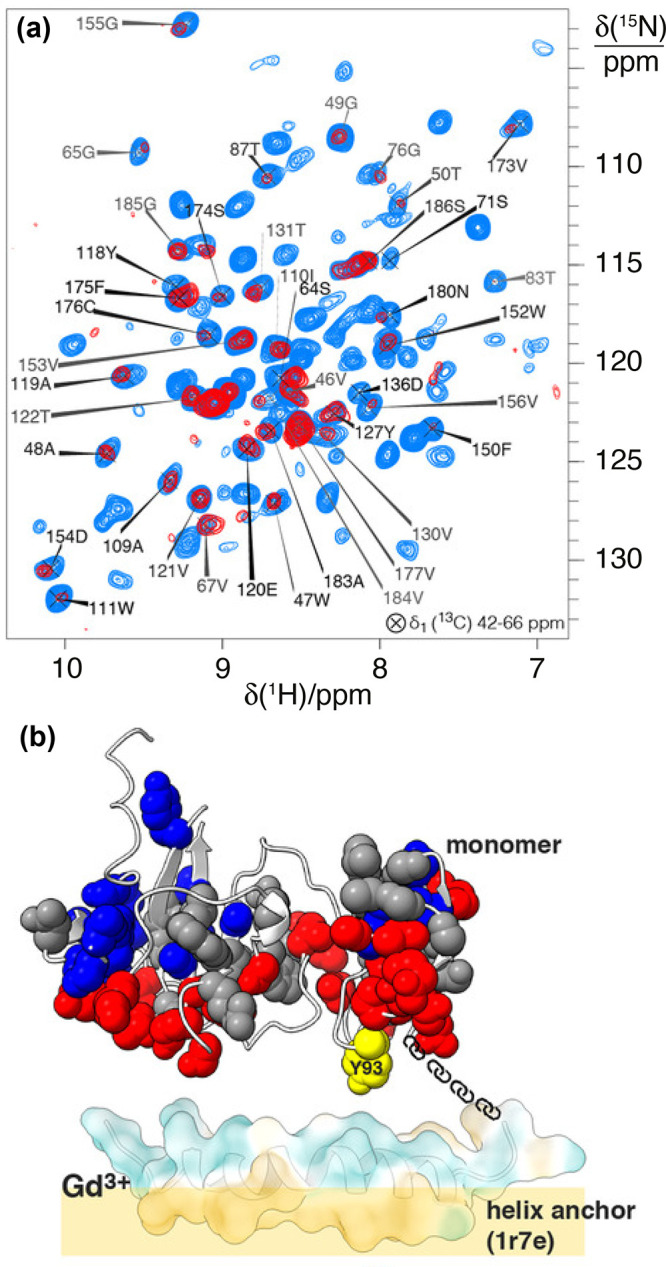
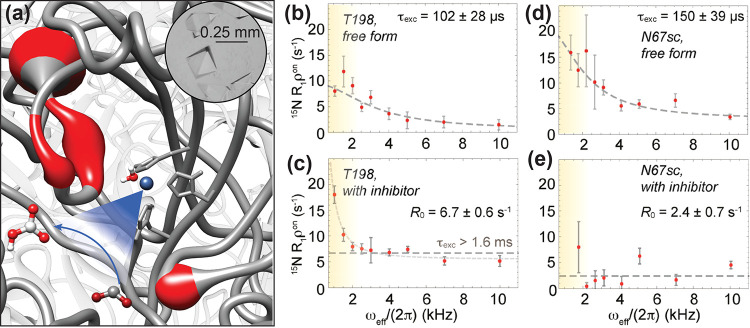
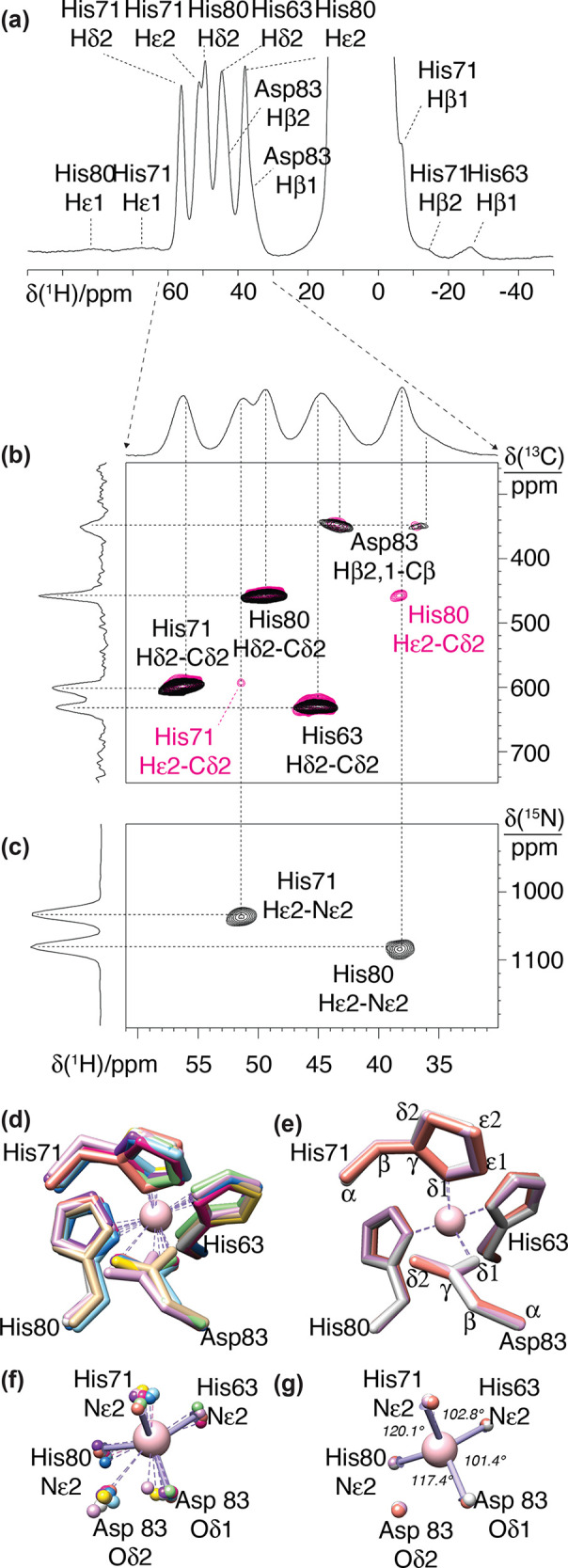
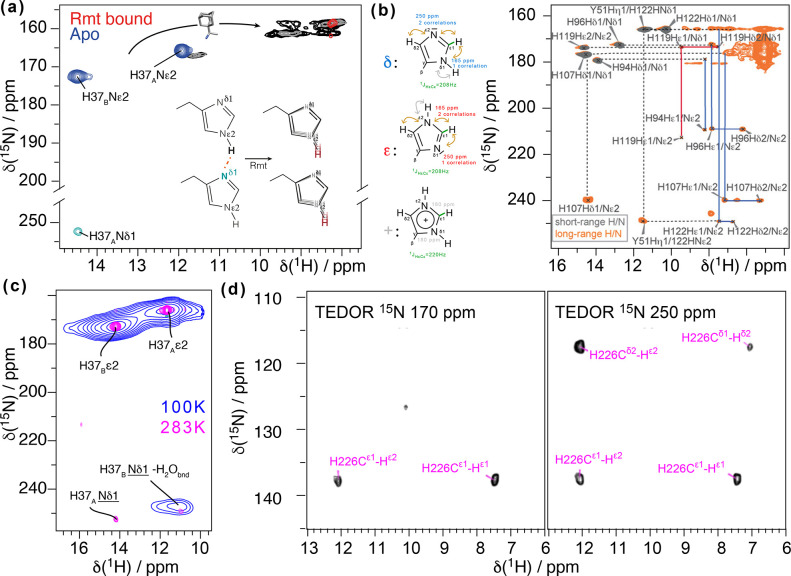
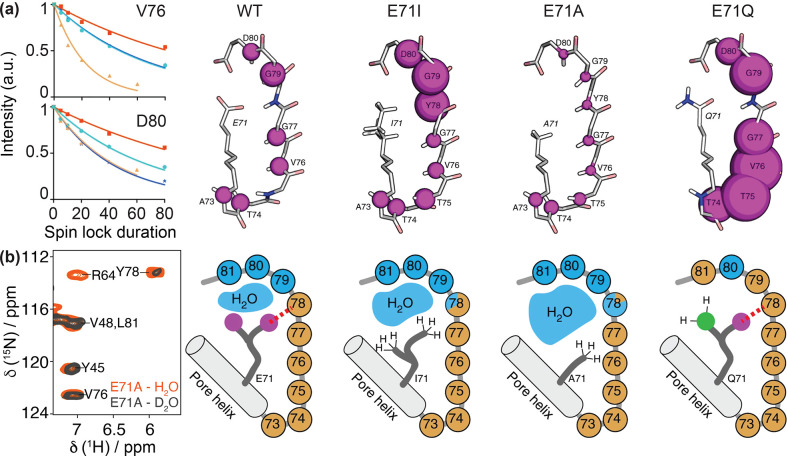
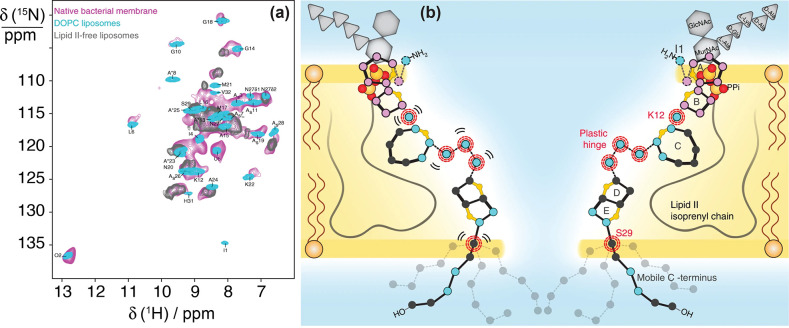
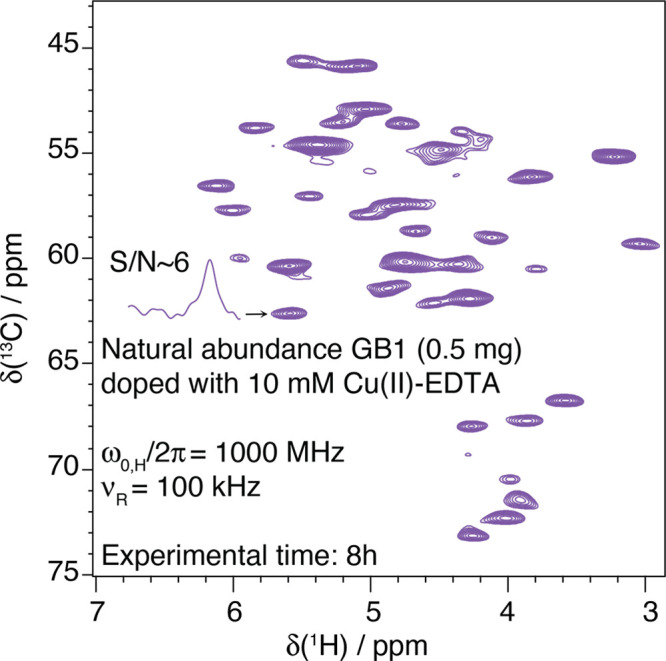
References
-
- Hologne M.; Chevelkov V.; Reif B. Deuterated Peptides and Proteins in MAS Solid-State NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 48, 211–232. 10.1016/j.pnmrs.2006.05.004. - DOI
-
- Reif B. In Protein NMR Techniques; Shekhtman A., Burz D. S., Eds.; Humana Press: Totowa, NJ, 2012; pp 279–301.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
