

NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition
- PMID: 35537449
- PMCID: PMC9520607
- DOI: 10.1016/j.molcel.2022.04.015
NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition
Abstract
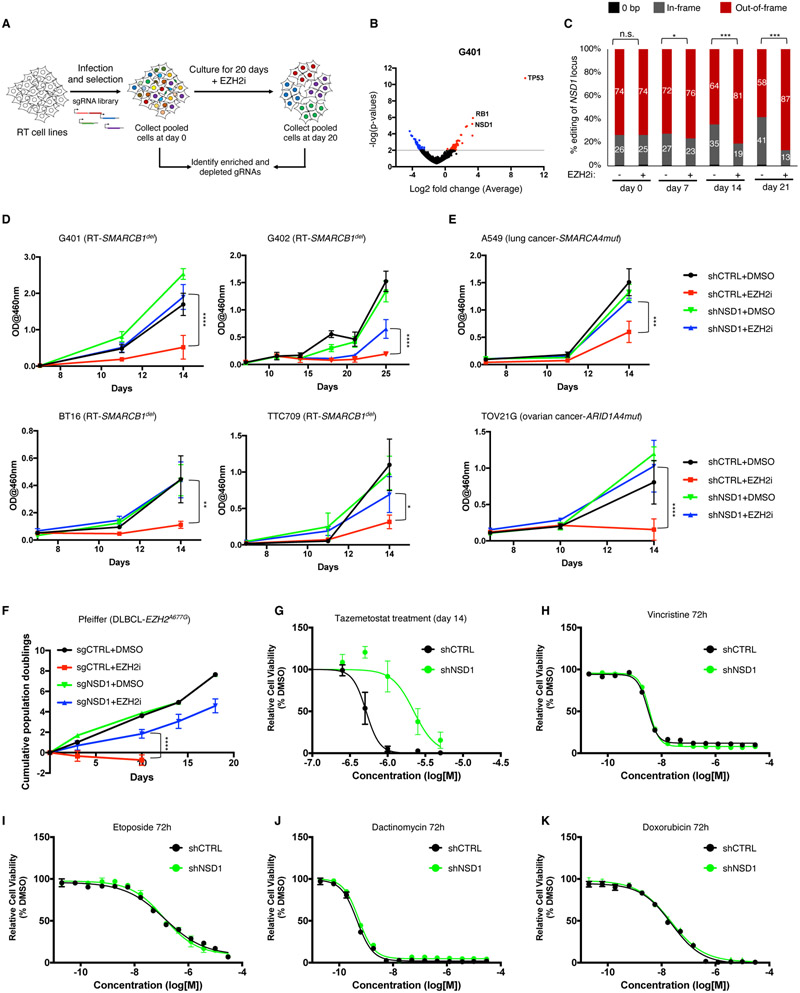
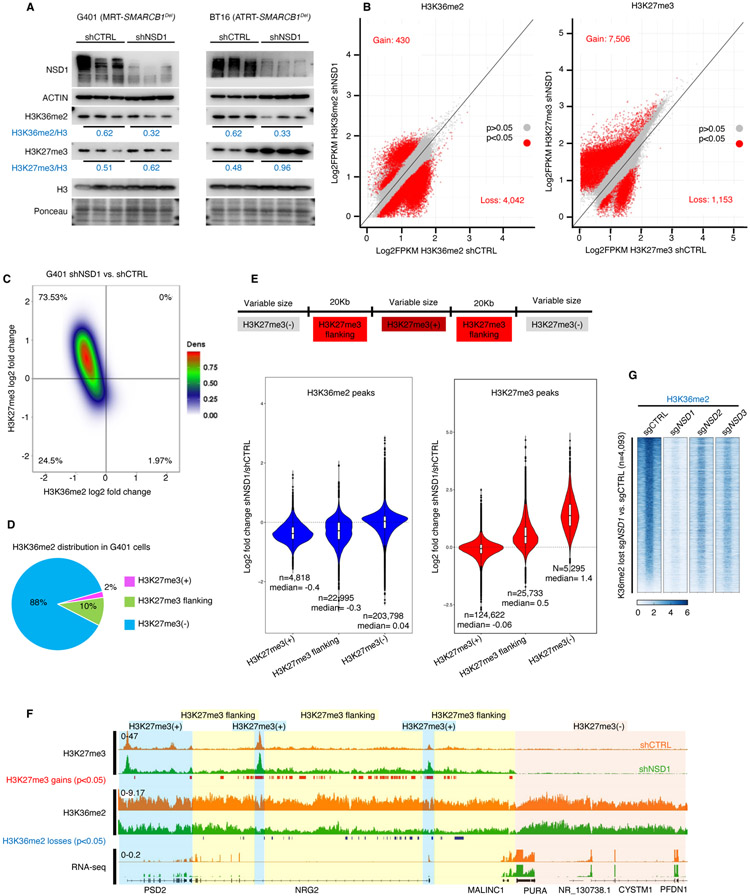
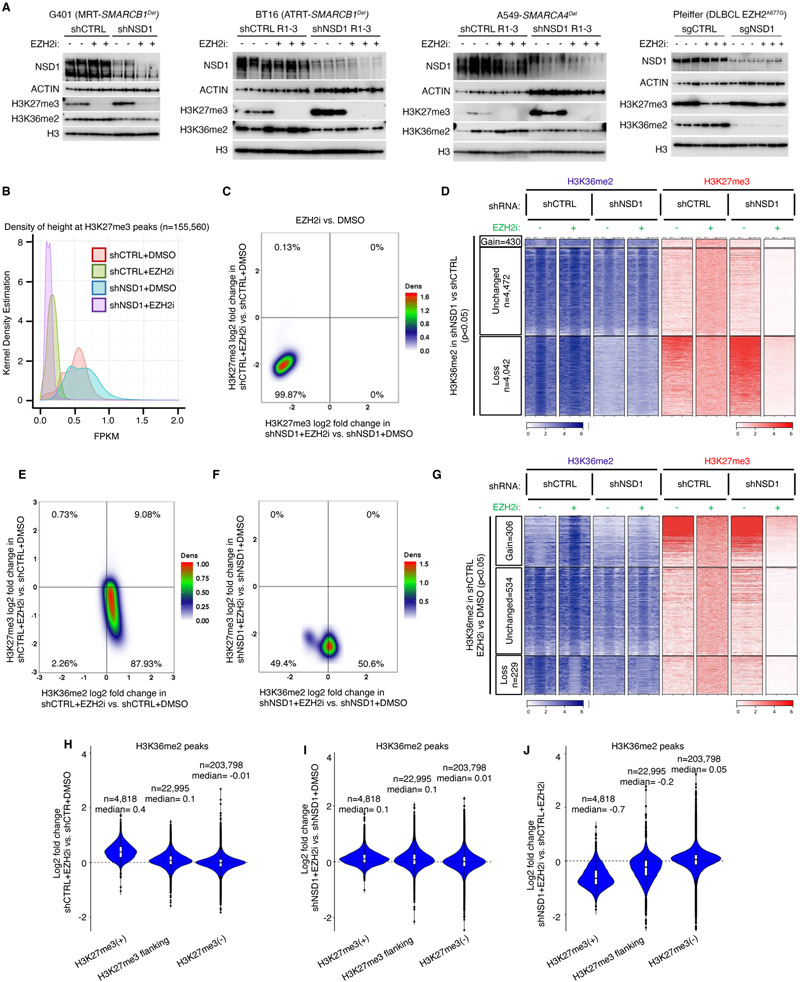
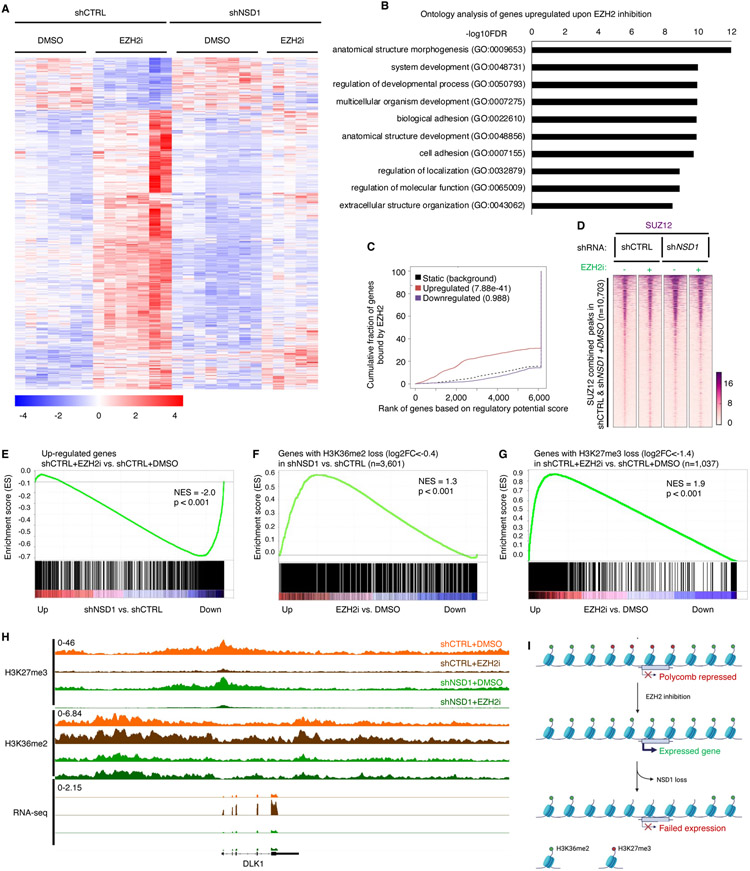
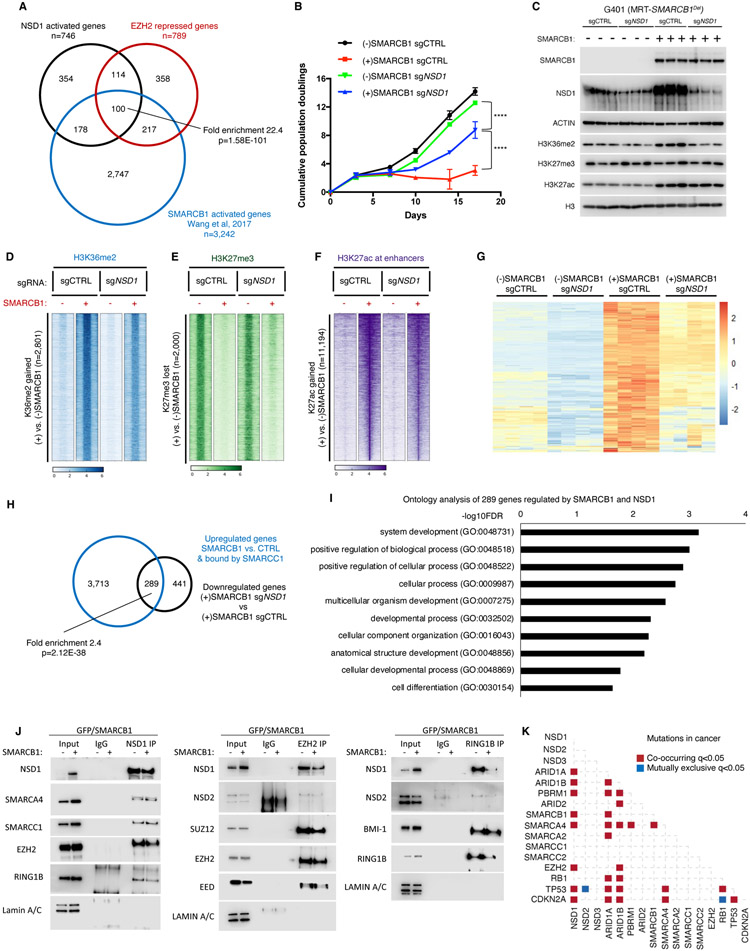
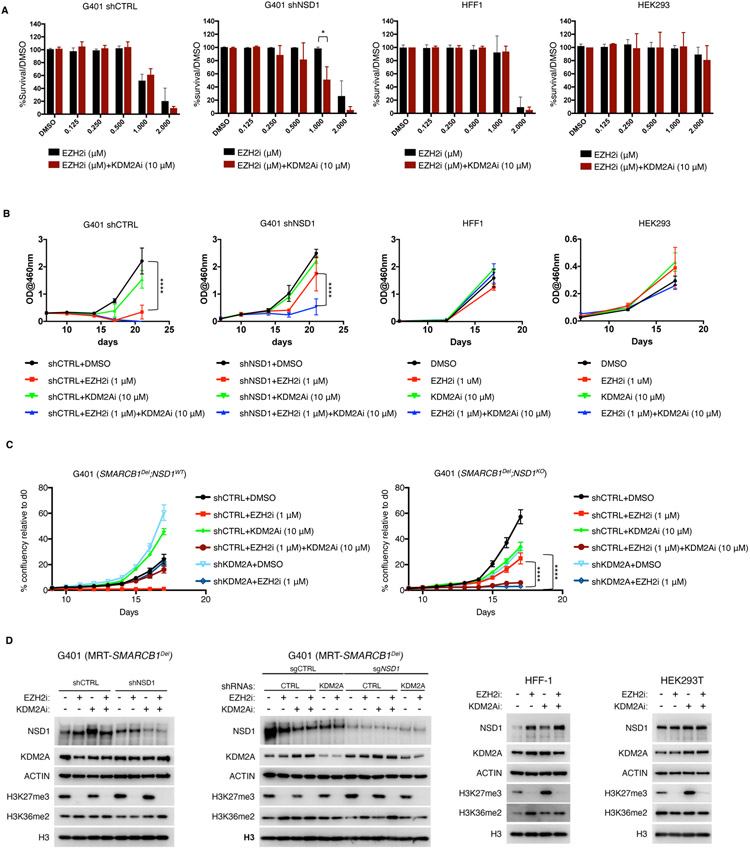
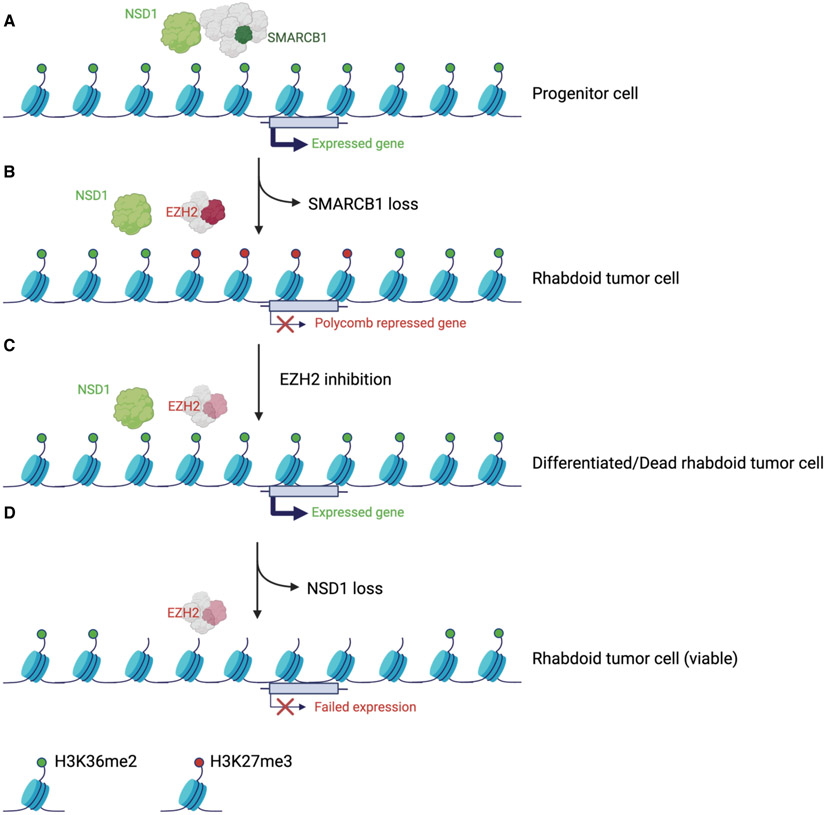
Disruption of antagonism between SWI/SNF chromatin remodelers and polycomb repressor complexes drives the formation of numerous cancer types. Recently, an inhibitor of the polycomb protein EZH2 was approved for the treatment of a sarcoma mutant in the SWI/SNF subunit SMARCB1, but resistance occurs. Here, we performed CRISPR screens in SMARCB1-mutant rhabdoid tumor cells to identify genetic contributors to SWI/SNF-polycomb antagonism and potential resistance mechanisms. We found that loss of the H3K36 methyltransferase NSD1 caused resistance to EZH2 inhibition. We show that NSD1 antagonizes polycomb via cooperation with SWI/SNF and identify co-occurrence of NSD1 inactivation in SWI/SNF-defective cancers, indicating in vivo relevance. We demonstrate that H3K36me2 itself has an essential role in the activation of polycomb target genes as inhibition of the H3K36me2 demethylase KDM2A restores the efficacy of EZH2 inhibition in SWI/SNF-deficient cells lacking NSD1. Together our data expand the mechanistic understanding of SWI/SNF and polycomb interplay and identify NSD1 as the key for coordinating this transcriptional control.
Keywords: BAF; EZH2 inhibition; NSD1; SWI/SNF; chromatin; epigenetics; polycomb; transcription.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Allis CD, and Jenuwein T (2016). The molecular hallmarks of epigenetic control. Nature Reviews Genetics 17, 487–500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
