

Endoplasmic reticulum stress mediates homocysteine-induced hypertrophy of cardiac cells through activation of cyclic nucleotide phosphodiesterase 1C
- PMID: 35538034
- PMCID: PMC9828163
- DOI: 10.3724/abbs.2022009
Endoplasmic reticulum stress mediates homocysteine-induced hypertrophy of cardiac cells through activation of cyclic nucleotide phosphodiesterase 1C
Abstract
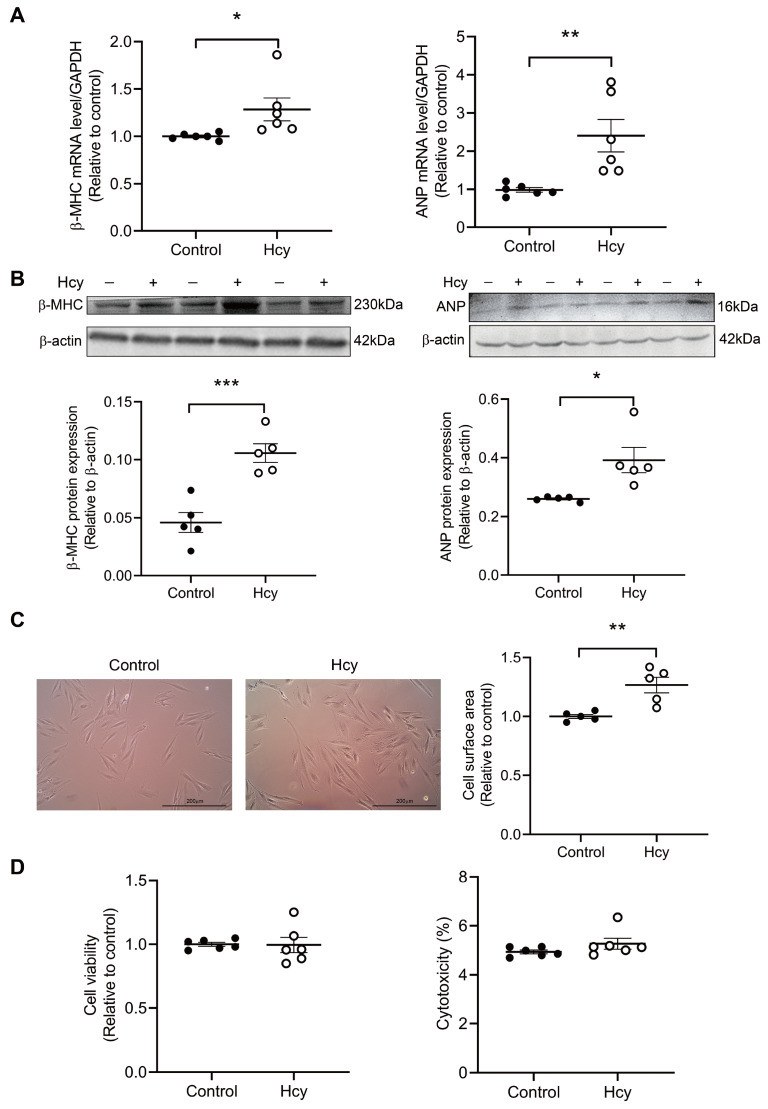
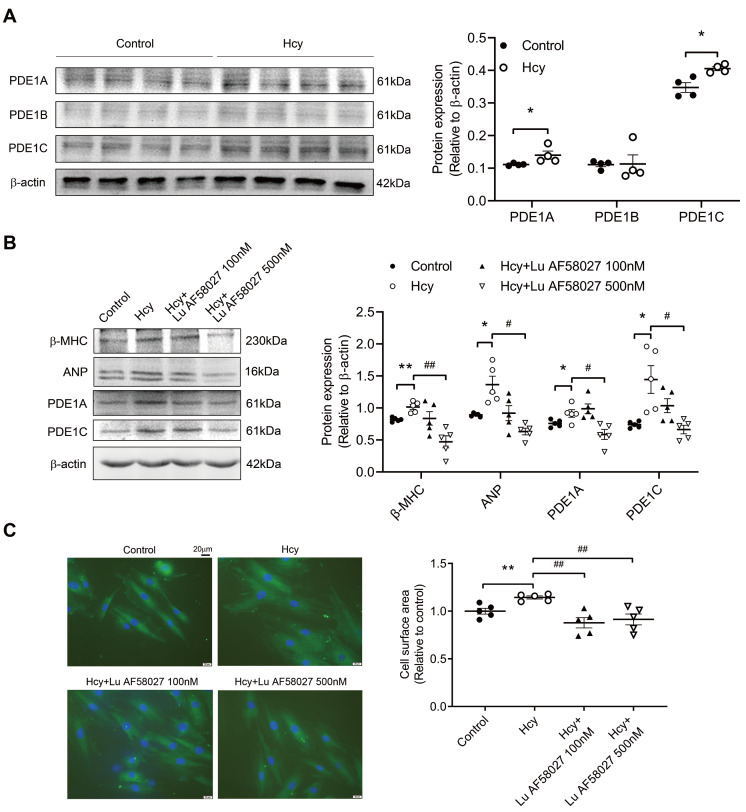
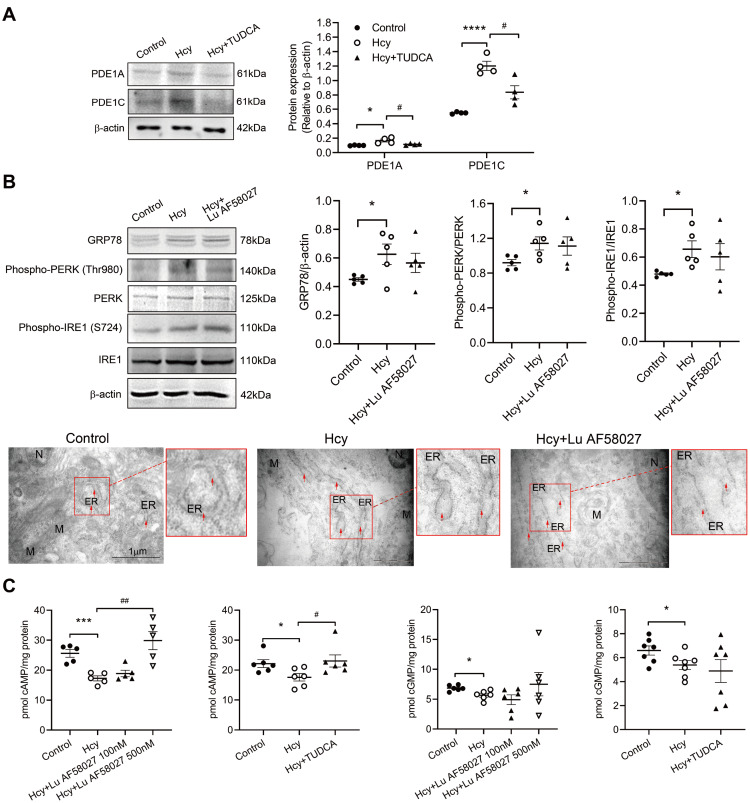
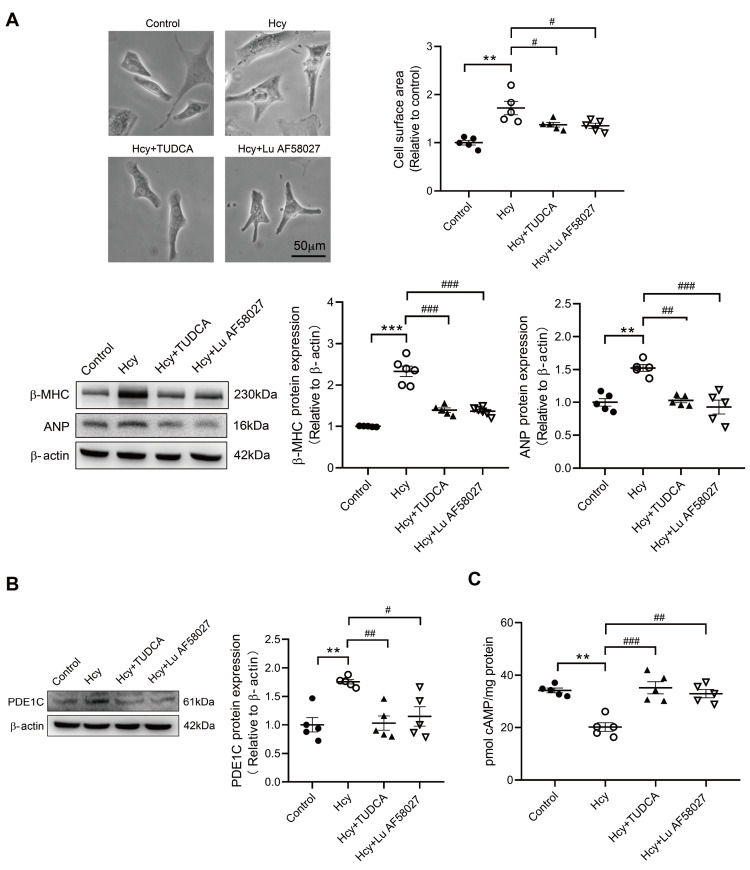
Although the association of elevated homocysteine level with cardiac hypertrophy has been reported, the molecular mechanisms by which homocysteine induces cardiac hypertrophy remain inadequately understood. In this study we aim to uncover the roles of cyclic nucleotide phosphodiesterase 1 (PDE1) and endoplasmic reticulum (ER) stress and their relationship to advance the mechanistic understanding of homocysteine-induced cardiac cell hypertrophy. H9c2 cells and primary neonatal rat cardiomyocytes are exposed to homocysteine with or without ER stress inhibitor TUDCA or PDE1-specific inhibitor Lu AF58027, or transfected with siRNAs targeting PDE1 isoforms prior to homocysteine-exposure. Cell surface area is measured and ultrastructure is examined by transmission electron microscopy. Hypertrophic markers, PDE1 isoforms, and ER stress molecules are detected by q-PCR and western blot analysis. Intracellular cGMP and cAMP are measured by ELISA. The results show that homocysteine causes the enlargement of H9c2 cells, increases the expressions of hypertrophic markers β-MHC and ANP, upregulates PDE1A and PDE1C, promotes the expressions of ER stress molecules, and causes ER dilatation and degranulation. TUDCA and Lu AF58027 downregulate β-MHC and ANP, and alleviate cell enlargement. TUDCA decreases PDE1A and PDE1C levels. Silencing of PDE1C inhibits homocysteine-induced hypertrophy, whereas PDE1A knockdown has minor effect. Both cAMP and cGMP are decreased after homocysteine-exposure, while only cAMP is restored by Lu AF58027 and TUDCA. TUDCA and Lu AF58027 also inhibit cell enlargement, downregulate ANP, β-MHC and PDE1C, and enhance cAMP level in homocysteine-exposed primary cardiomyocytes. ER stress mediates homocysteine-induced hypertrophy of cardiac cells via upregulating PDE1C expression Cyclic nucleotide, especially cAMP, is the downstream mediator of the ER stress-PDE1C signaling axis in homocysteine-induced cell hypertrophy.
Keywords: cardiac hypertrophy; endoplasmic reticulum stress; homocysteine; phosphodiesterase.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


References
-
- Cao Z, Zhang Y, Sun T, Zhang S, Yu W, Zhu J. Homocysteine induces cardiac hypertrophy by up-regulating ATP7a expression. Int J Clin Exp Pathol. 2015, 8: 12829–12836 - PMC - PubMed
-
- Kar S, Shahshahan HR, Kambis TN, Yadav SK, Li Z, Lefer DJ, Mishra PK. Hydrogen sulfide ameliorates homocysteine-induced cardiac remodeling and dysfunction. Front Physiol. . 2019;10:598. doi: 10.3389/fphys.2019.00598. - DOI - PMC - PubMed
-
- Kassab S, Garadah T, Abu-Hijleh M, Golbahar J, Senok S, Wazir J, Gumaa K. The angiotensin type 1 receptor antagonist valsartan attenuates pathological ventricular hypertrophy induced by hyperhomocysteinemia in rats. J Renin Angiotensin Aldosterone Syst. . 2006;7:206–211. doi: 10.3317/jraas.2006.039. - DOI - PubMed
-
- Raaf L, Noll C, Cherifi MEH, Samuel JL, Delcayre C, Delabar JM, Benazzoug Y, et al. Myocardial fibrosis and TGFB expression in hyperhomocysteinemic rats. Mol Cell Biochem. . 2011;347:63–70. doi: 10.1007/s11010-010-0612-5. - DOI - PubMed
-
- Sipkens JA, Krijnen PA, Meischl C, Cillessen SA, Smulders YM, Smith DE, Giroth CP, et al. Homocysteine affects cardiomyocyte viability: concentration-dependent effects on reversible flip-flop, apoptosis and necrosis. Apoptosis. . 2007;12:1407–1418. doi: 10.1007/s10495-007-0077-5. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
