

The genomic and transcriptional landscape of primary central nervous system lymphoma
- PMID: 35538064
- PMCID: PMC9091224
- DOI: 10.1038/s41467-022-30050-y
The genomic and transcriptional landscape of primary central nervous system lymphoma
Abstract
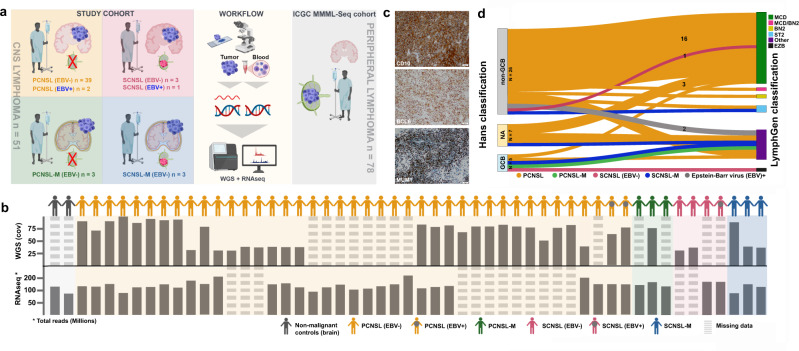
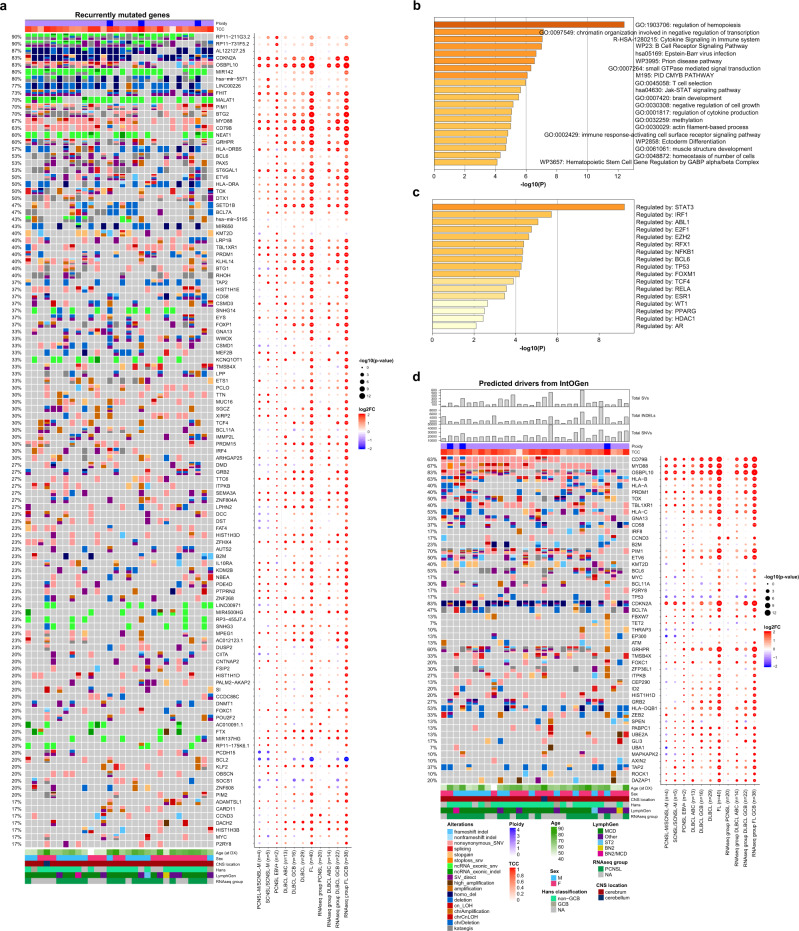
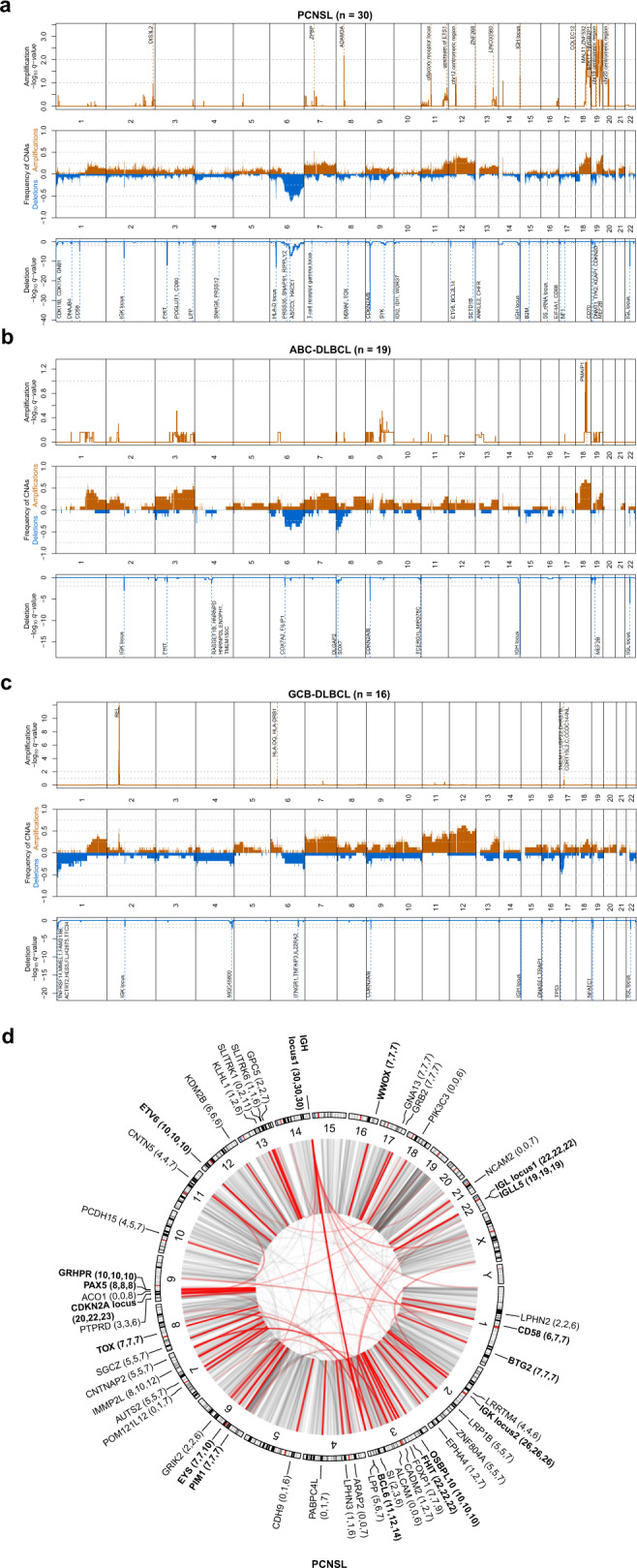
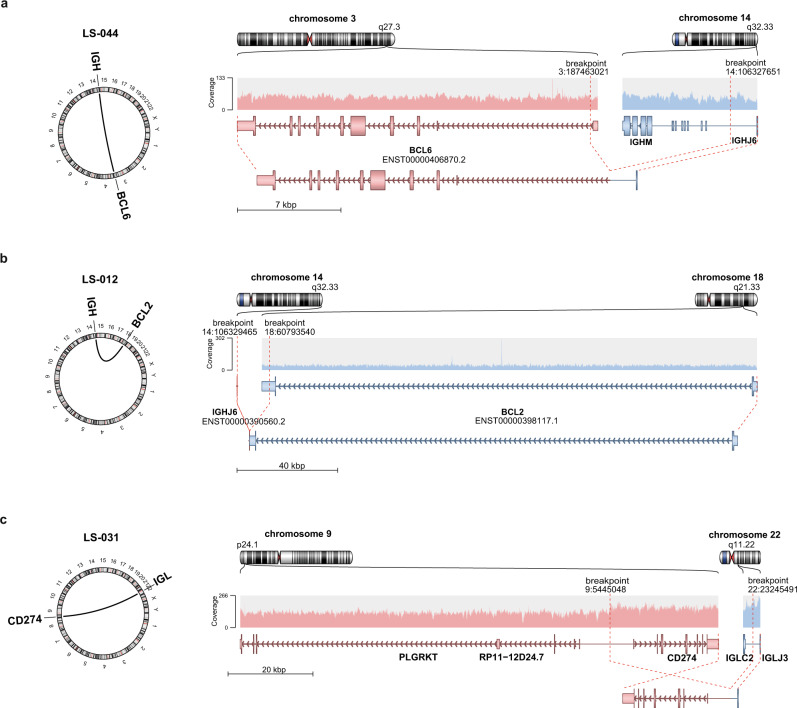
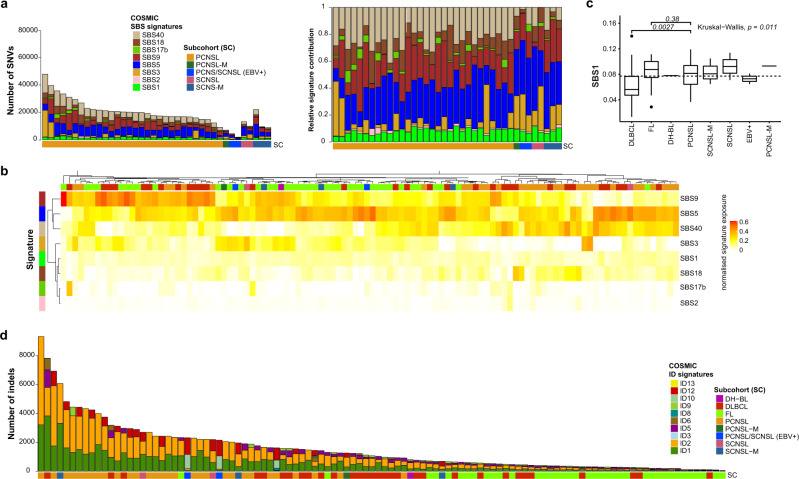
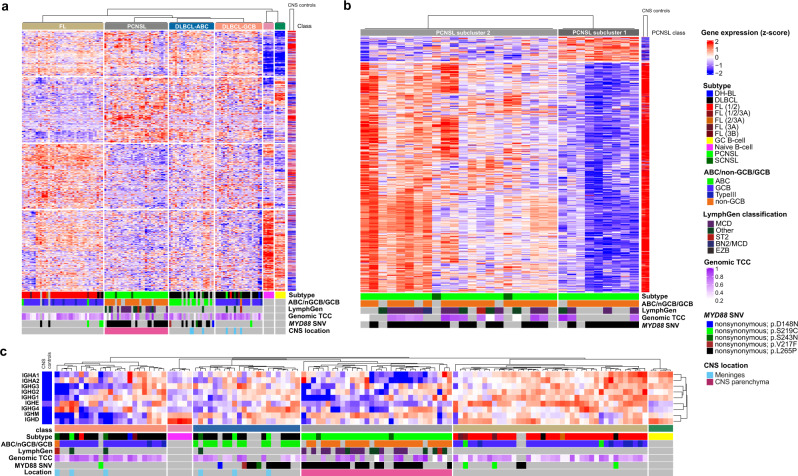
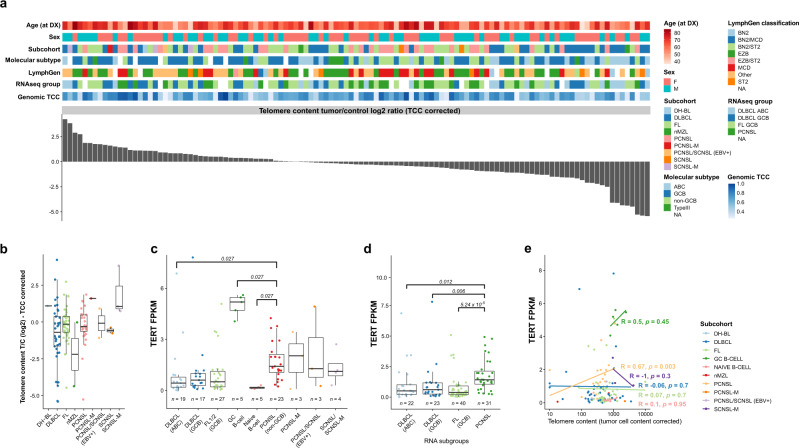
Primary lymphomas of the central nervous system (PCNSL) are mainly diffuse large B-cell lymphomas (DLBCLs) confined to the central nervous system (CNS). Molecular drivers of PCNSL have not been fully elucidated. Here, we profile and compare the whole-genome and transcriptome landscape of 51 CNS lymphomas (CNSL) to 39 follicular lymphoma and 36 DLBCL cases outside the CNS. We find recurrent mutations in JAK-STAT, NFkB, and B-cell receptor signaling pathways, including hallmark mutations in MYD88 L265P (67%) and CD79B (63%), and CDKN2A deletions (83%). PCNSLs exhibit significantly more focal deletions of HLA-D (6p21) locus as a potential mechanism of immune evasion. Mutational signatures correlating with DNA replication and mitosis are significantly enriched in PCNSL. TERT gene expression is significantly higher in PCNSL compared to activated B-cell (ABC)-DLBCL. Transcriptome analysis clearly distinguishes PCNSL and systemic DLBCL into distinct molecular subtypes. Epstein-Barr virus (EBV)+ CNSL cases lack recurrent mutational hotspots apart from IG and HLA-DRB loci. We show that PCNSL can be clearly distinguished from DLBCL, having distinct expression profiles, IG expression and translocation patterns, as well as specific combinations of genetic alterations.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Louis, D. N., Ohgaki, H., Wiestler, O. D. & Cavenee, W. K. WHO Classification of Tumours of the Central Nervous System 4 edn, 1 (International Agency for Research on Cancer Lyon, 2016).
-
- Swerdlow, S. H. et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues 2 (International Agency for Research on Cancer Lyon, 2008).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
