

Recombineering and MAGE
- PMID: 35540496
- PMCID: PMC9083505
- DOI: 10.1038/s43586-020-00006-x
Recombineering and MAGE
Abstract
Recombination-mediated genetic engineering, also known as recombineering, is the genomic incorporation of homologous single-stranded or double-stranded DNA into bacterial genomes. Recombineering and its derivative methods have radically improved genome engineering capabilities, perhaps none more so than multiplex automated genome engineering (MAGE). MAGE is representative of a set of highly multiplexed single-stranded DNA-mediated technologies. First described in Escherichia coli, both MAGE and recombineering are being rapidly translated into diverse prokaryotes and even into eukaryotic cells. Together, this modern set of tools offers the promise of radically improving the scope and throughput of experimental biology by providing powerful new methods to ease the genetic manipulation of model and non-model organisms. In this Primer, we describe recombineering and MAGE, their optimal use, their diverse applications and methods for pairing them with other genetic editing tools. We then look forward to the future of genetic engineering.
Conflict of interest statement
Competing interests T.M.W, G.T.F. and G.M.C. are inventors on a patent application related to serial enrichment for efficient recombineering (SEER) and new single-stranded DNA-annealing protein (SSAP) discovery. A.N. and C.P. are inventors on a patent related to directed evolution with random genomic mutations (DIvERGE) (US10669537B2: Mutagenizing Intracellular Nucleic Acids). F.J.I. and G.M.C. are inventors on a MAGE patent, which has been licensed. F.J.I. is an inventor on a patent application related to eukaryotic MAGE. The remaining authors declare no competing interests.
Figures
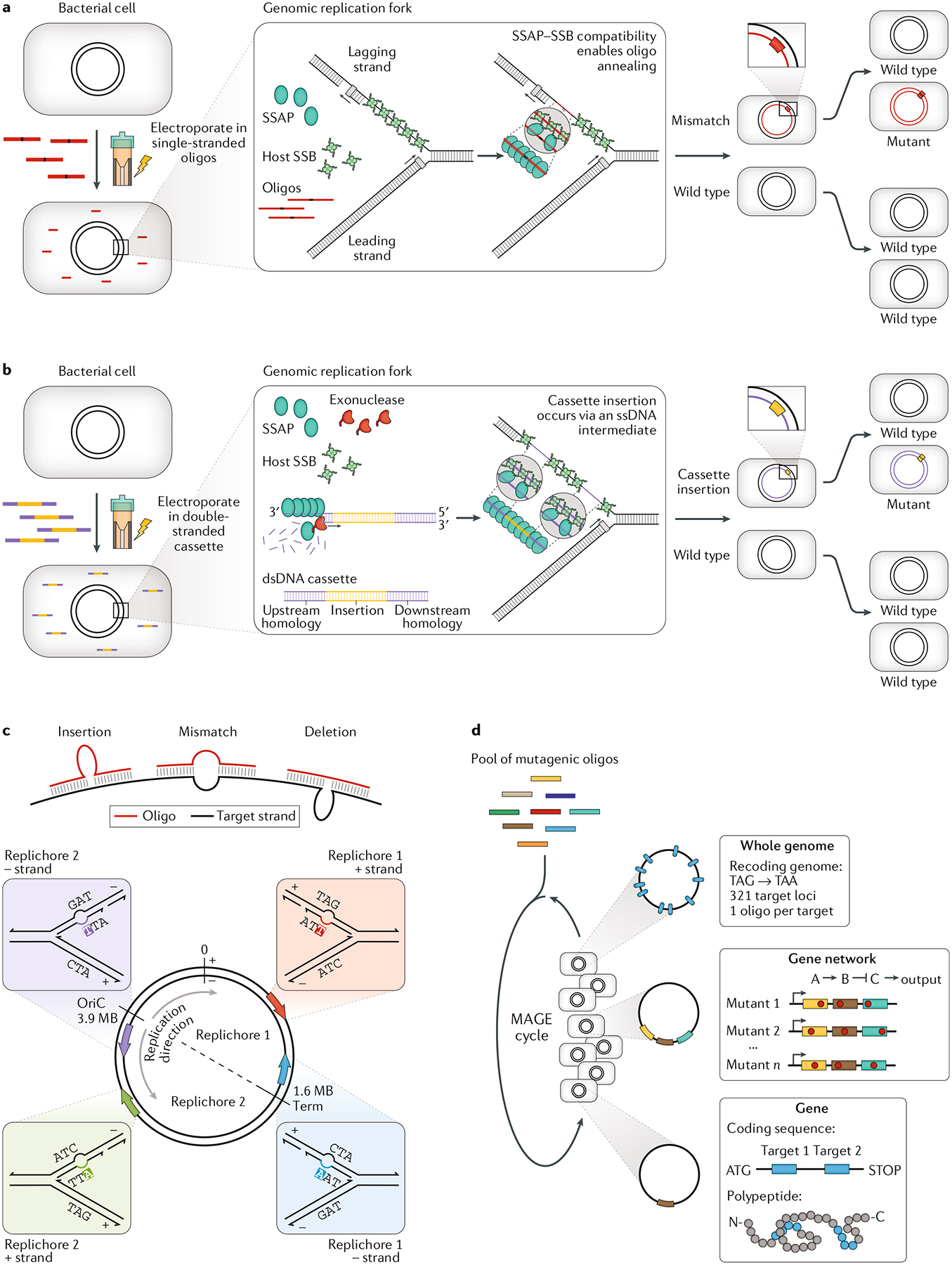

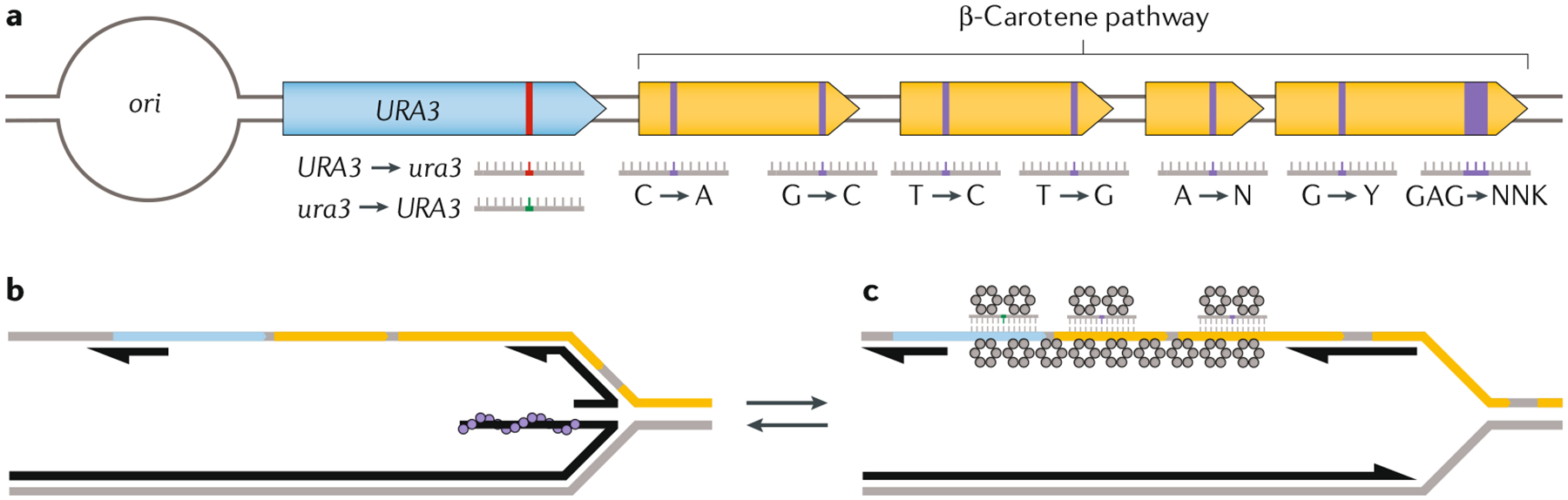
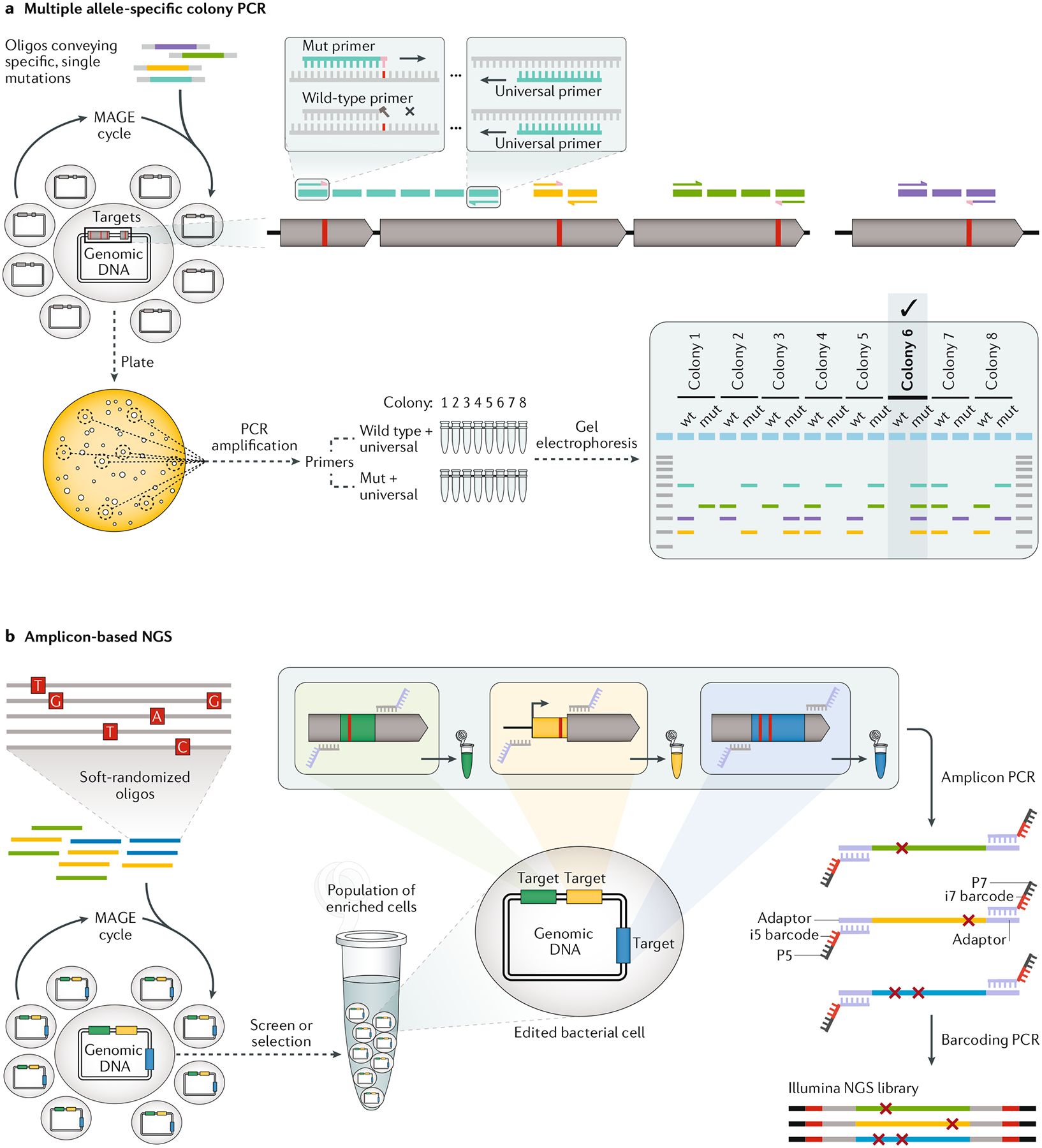
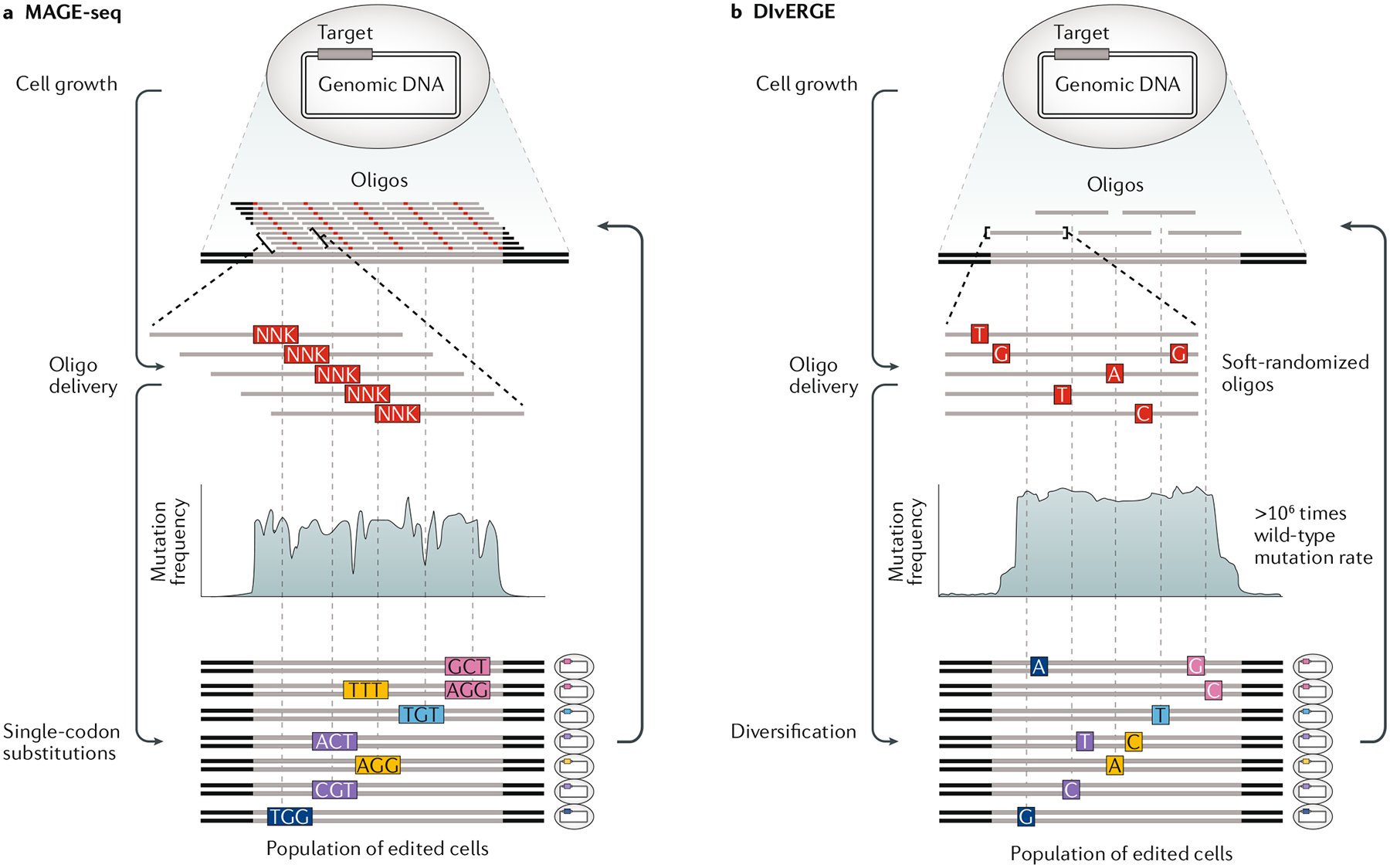


References
-
- Simon R, Priefer U & Pühler A A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol 1, 784–791 (1983).
-
- Ye B et al. Unmarked genetic manipulation in Bacillus subtilis by natural co-transformation. J. Biotechnol 284, 57–62 (2018). - PubMed
-
- Doudna JA & Charpentier E Genome editing. The new frontier of genome engineering with CRISPR–Cas9. Science 346, 1258096 (2014). - PubMed
-
- Gersbach CA Genome engineering: the next genomic revolution. Nat. Methods 11, 1009–1011 (2014). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources