

Introducing DDEC6 atomic population analysis: part 4. Efficient parallel computation of net atomic charges, atomic spin moments, bond orders, and more
- PMID: 35541489
- PMCID: PMC9077577
- DOI: 10.1039/c7ra11829e
Introducing DDEC6 atomic population analysis: part 4. Efficient parallel computation of net atomic charges, atomic spin moments, bond orders, and more
Abstract
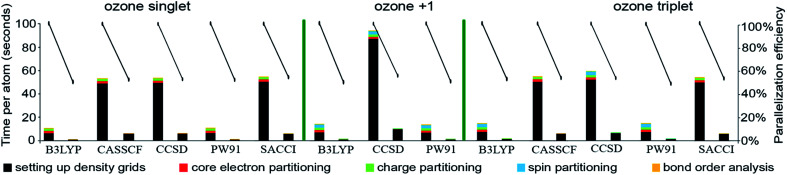
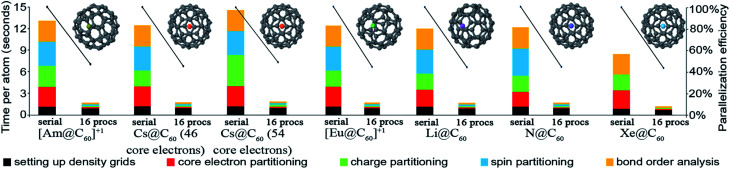
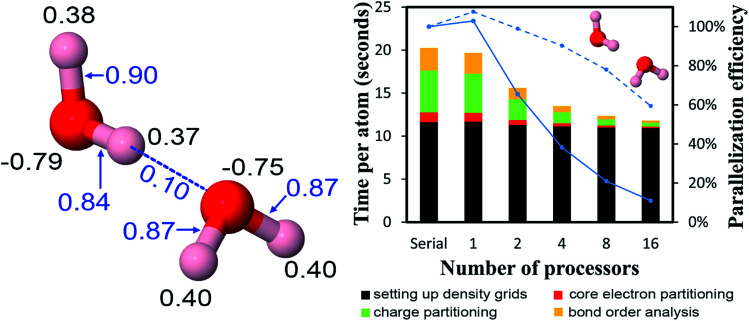
The DDEC6 method is one of the most accurate and broadly applicable atomic population analysis methods. It works for a broad range of periodic and non-periodic materials with no magnetism, collinear magnetism, and non-collinear magnetism irrespective of the basis set type. First, we show DDEC6 charge partitioning to assign net atomic charges corresponds to solving a series of 14 Lagrangians in order. Then, we provide flow diagrams for overall DDEC6 analysis, spin partitioning, and bond order calculations. We wrote an OpenMP parallelized Fortran code to provide efficient computations. We show that by storing large arrays as shared variables in cache line friendly order, memory requirements are independent of the number of parallel computing cores and false sharing is minimized. We show that both total memory required and the computational time scale linearly with increasing numbers of atoms in the unit cell. Using the presently chosen uniform grids, computational times of ∼9 to 94 seconds per atom were required to perform DDEC6 analysis on a single computing core in an Intel Xeon E5 multi-processor unit. Parallelization efficiencies were usually >50% for computations performed on 2 to 16 cores of a cache coherent node. As examples we study a B-DNA decamer, nickel metal, supercells of hexagonal ice crystals, six X@C60 endohedral fullerene complexes, a water dimer, a Mn12-acetate single molecule magnet exhibiting collinear magnetism, a Fe4O12N4C40H52 single molecule magnet exhibiting non-collinear magnetism, and several spin states of an ozone molecule. Efficient parallel computation was achieved for systems containing as few as one and as many as >8000 atoms in a unit cell. We varied many calculation factors (e.g., grid spacing, code design, thread arrangement, etc.) and report their effects on calculation speed and precision. We make recommendations for excellent performance.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts of interest to declare.
Figures




















References
-
- Erucar I. Manz T. A. Keskin S. Mol. Simul. 2014;40:557–570. doi: 10.1080/08927022.2013.829219. - DOI
-
- Gates T. S. Odegard G. M. Frankland S. J. V. Clancy T. C. Compos. Sci. Technol. 2005;65:2416–2434. doi: 10.1016/j.compscitech.2005.06.009. - DOI
-
- Spivey J. J. Krishna K. S. Kumar C. S. S. R. Dooley K. M. Flake J. C. Haber L. H. Xu Y. Janik M. J. Sinnott S. B. Cheng Y. T. Liang T. Sholl D. S. Manz T. A. Diebold U. Parkinson G. S. Bruce D. A. de Jongh P. J. Phys. Chem. C. 2014;118:20043–20069.
LinkOut - more resources
Full Text Sources
Research Materials