

DOCKopathies: A systematic review of the clinical pathologies associated with human DOCK pathogenic variants
- PMID: 35544951
- PMCID: PMC9357139
- DOI: 10.1002/humu.24398
DOCKopathies: A systematic review of the clinical pathologies associated with human DOCK pathogenic variants
Abstract
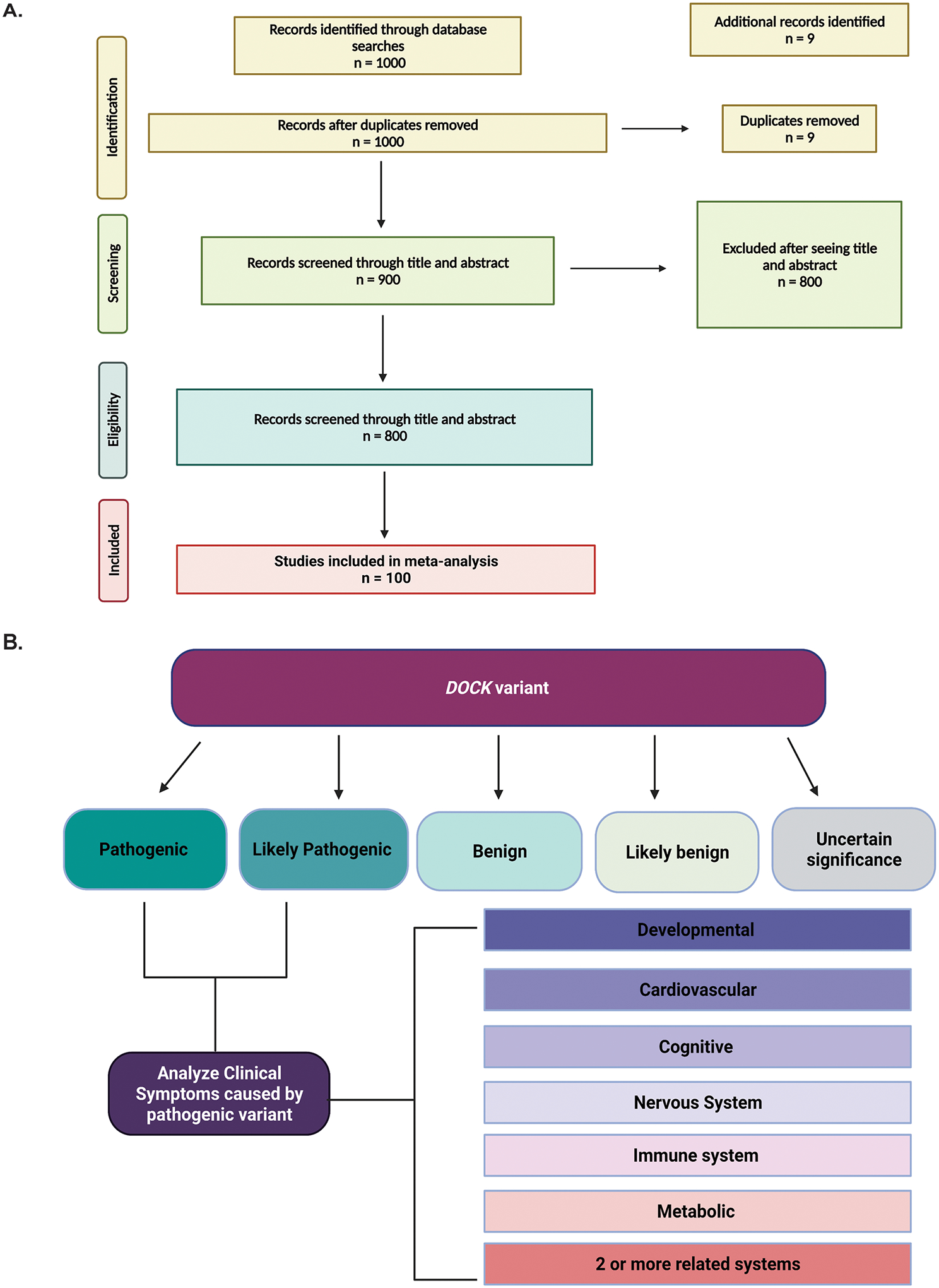
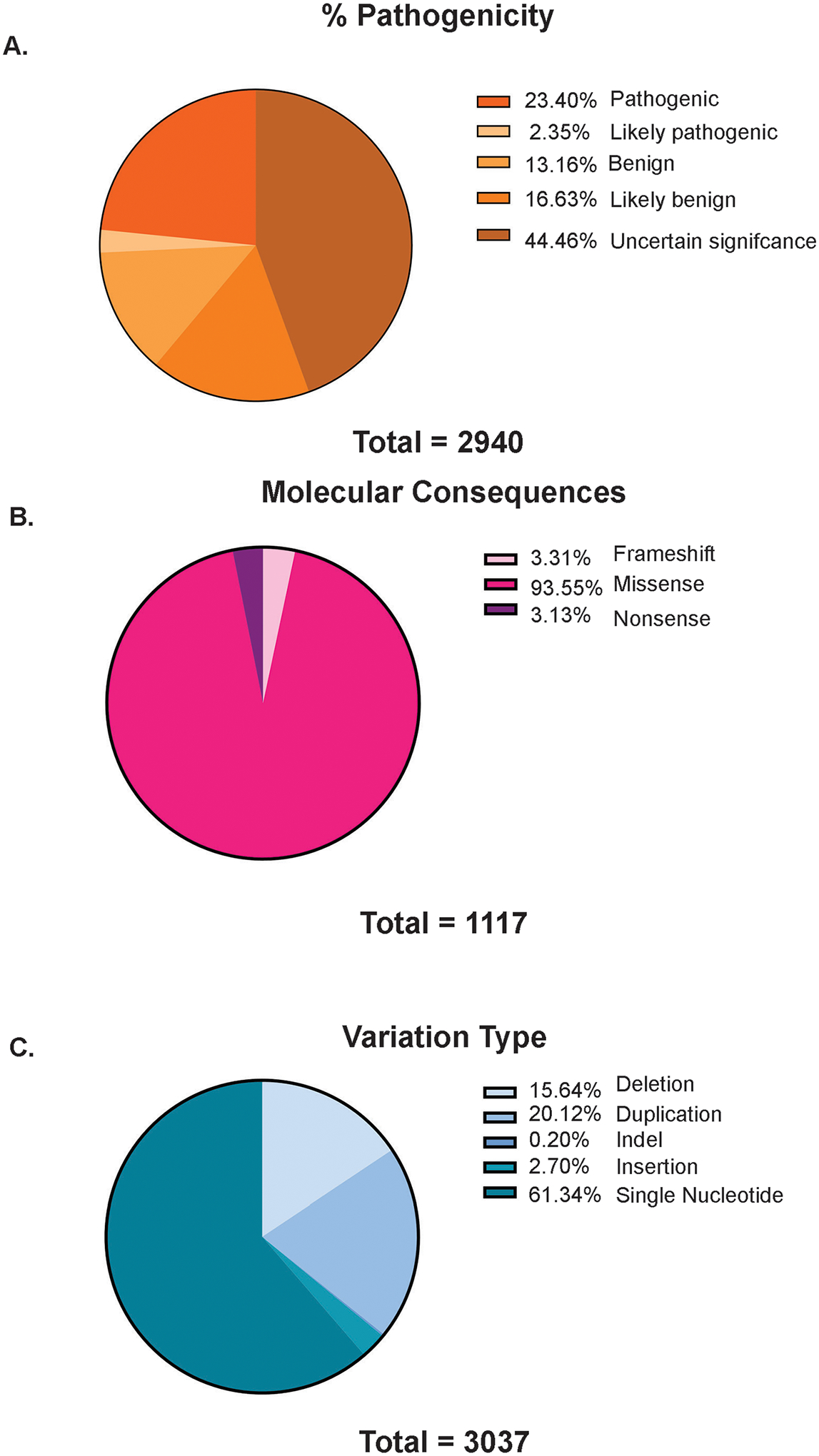
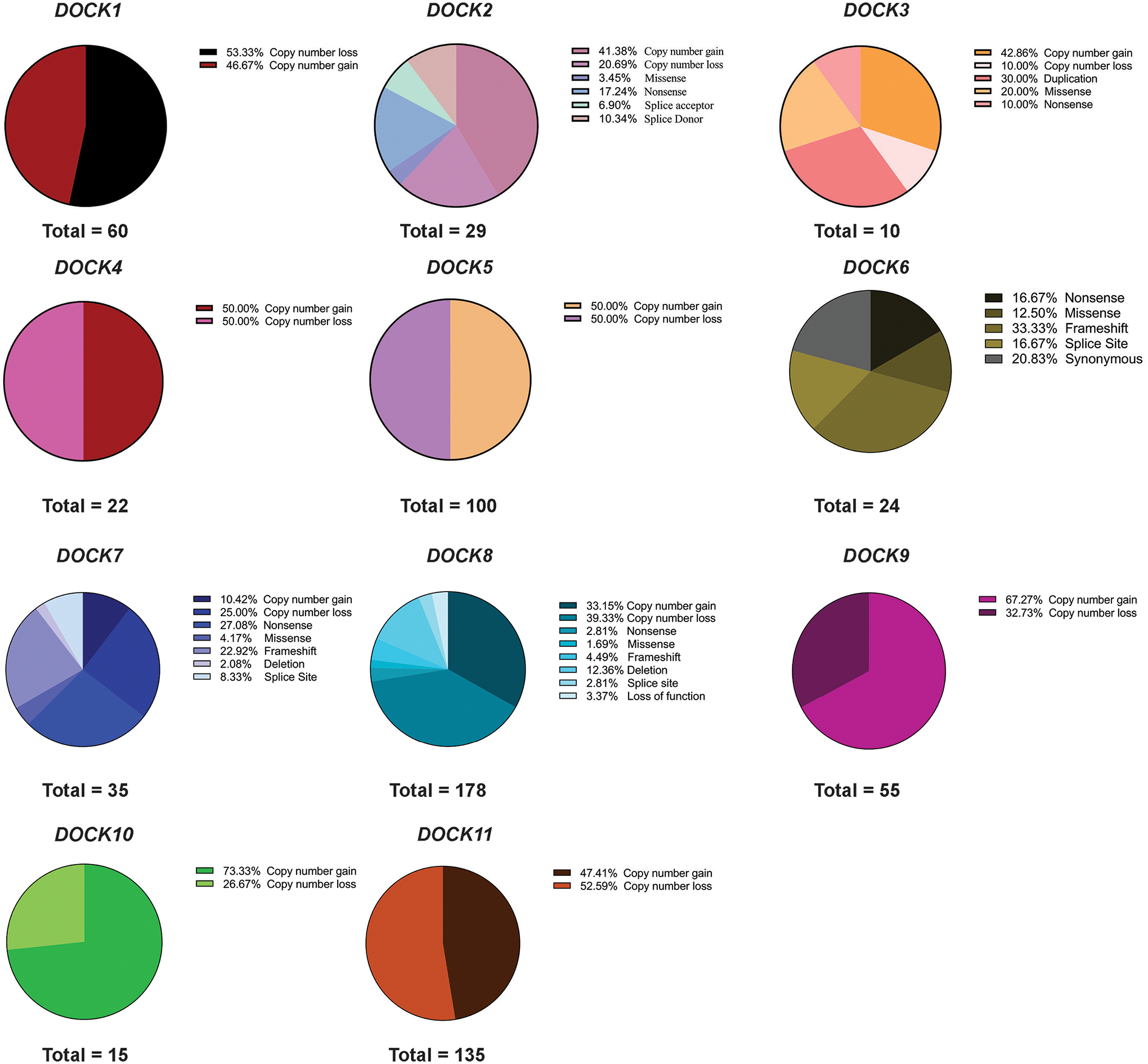
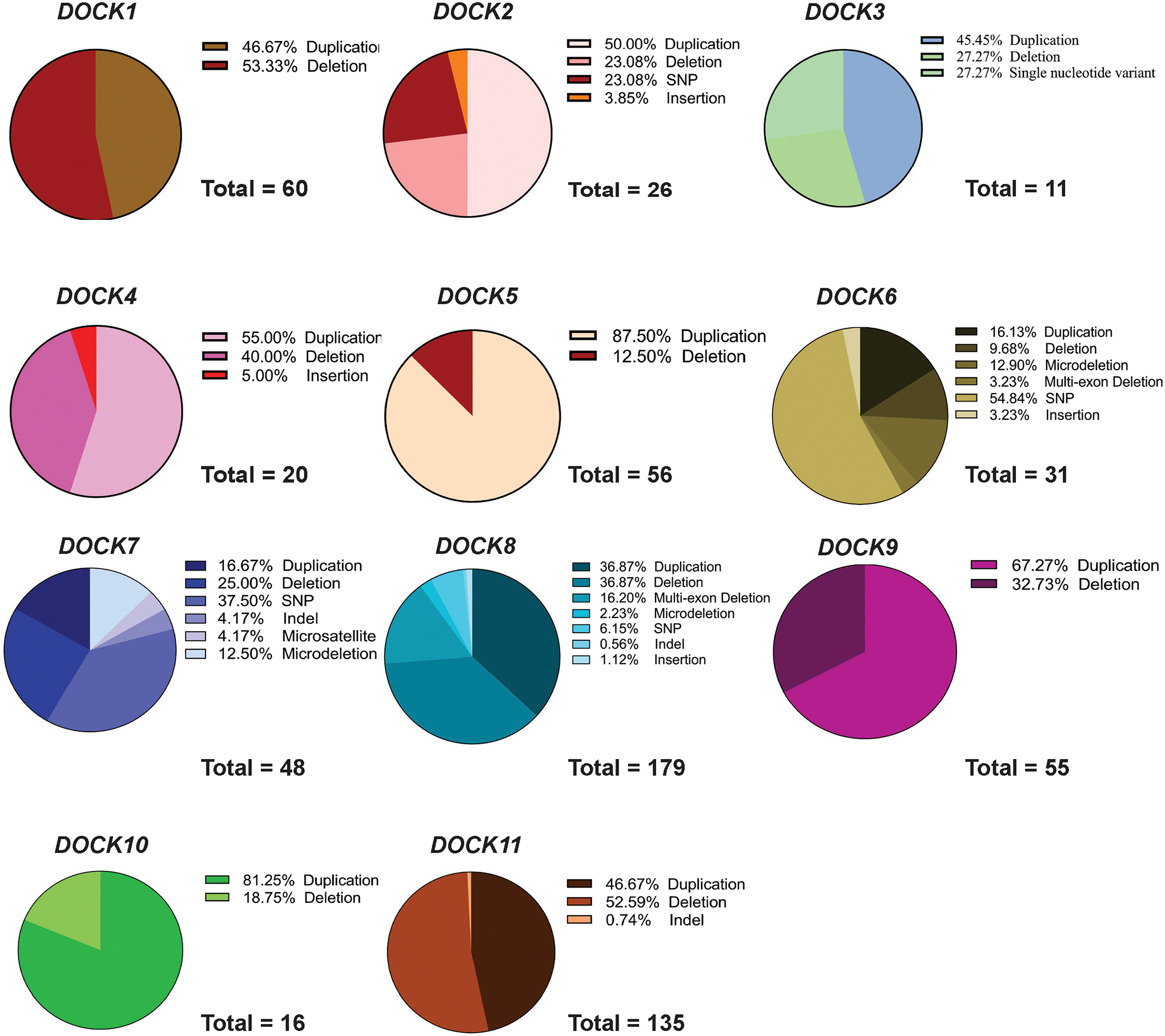
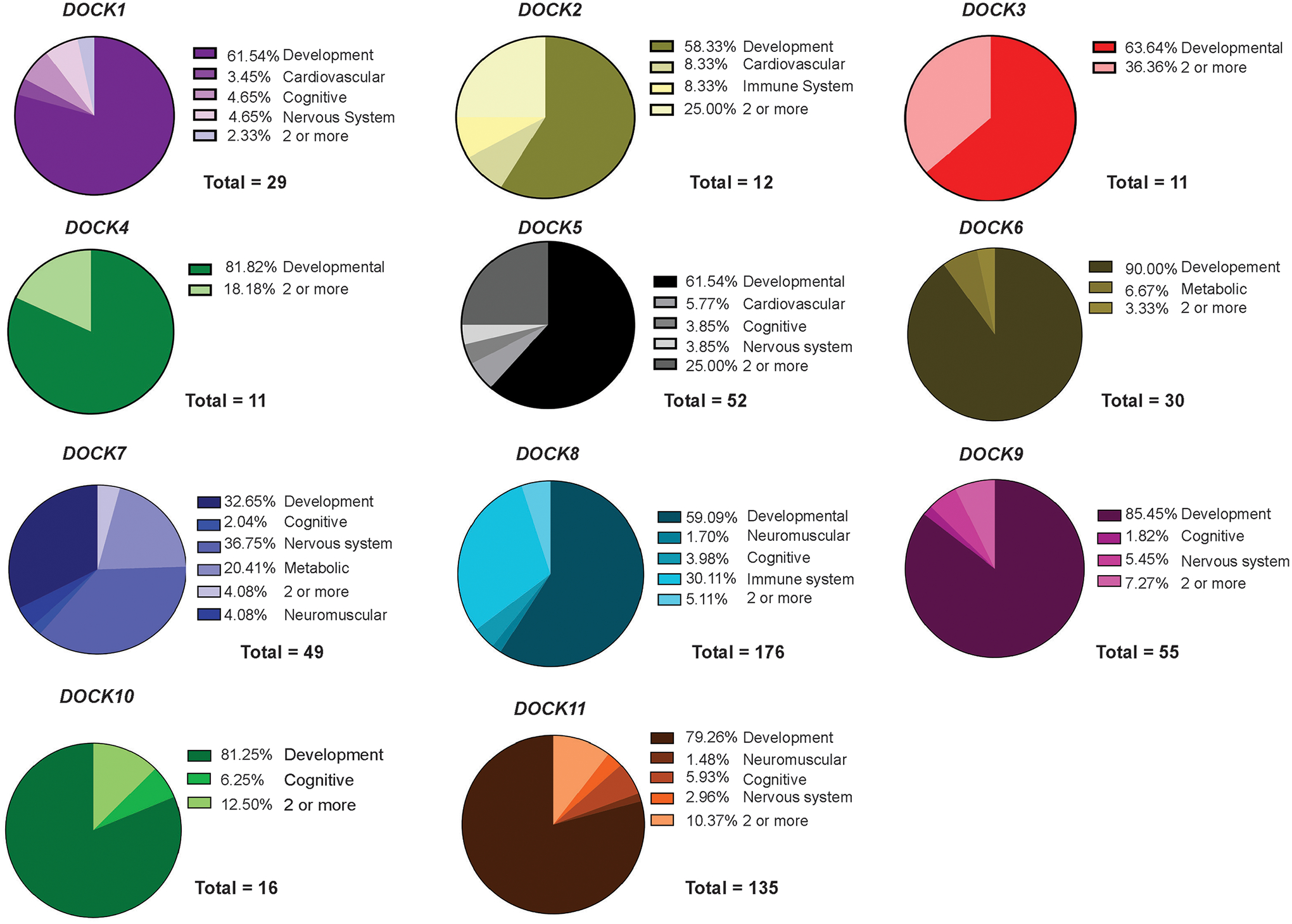
The Dedicator of Cytokinesis (DOCK) family (DOCK1-11) of genes are essential mediators of cellular migration, growth, and fusion in a variety of cell types and tissues. Recent advances in whole-genome sequencing of patients with undiagnosed genetic disorders have identified several rare pathogenic variants in DOCK genes. We conducted a systematic review and performed a patient database and literature search of reported DOCK pathogenic variants that have been identified in association with clinical pathologies such as global developmental delay, immune cell dysfunction, muscle hypotonia, and muscle ataxia among other categories. We then categorized these pathogenic DOCK variants and their associated clinical phenotypes under several unique categories: developmental, cardiovascular, metabolic, cognitive, or neuromuscular. Our systematic review of DOCK variants aims to identify and analyze potential DOCK-regulated networks associated with neuromuscular diseases and other disease pathologies, which may identify novel therapeutic strategies and targets. This systematic analysis and categorization of human-associated pathologies with DOCK pathogenic variants is the first report to the best of our knowledge for a unique class in this understudied gene family that has important implications in furthering personalized genomic medicine, clinical diagnoses, and improve targeted therapeutic outcomes across many clinical pathologies.
Keywords: DOCK; hypotonia; intellectual disability; skeletal muscle.
© 2022 Wiley Periodicals LLC.
Conflict of interest statement
Figures
References
Web resources:
References
-
- Aartsma-Rus A, Verschuuren J, Campion G, van Ommen G. j., & van Deutekom J (2012). Exon skipping for DMD. Orphanet Journal of Rare Diseases, 7(Suppl 2), A20. http://www.ojrd.com/content/7/S2/A20
-
- Akcakaya P, Bobbin ML, Guo JA, Malagon-Lopez J, Clement K, Garcia SP, Fellows MD, Porritt MJ, Firth MA, Carreras A, Baccega T, Seeliger F, Bjursell M, Tsai SQ, Nguyen NT, Nitsch R, Mayr LM, Pinello L, Bohlooly-Y M, Aryee MJ, Maresca M, & Joung JK (2018). In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 10.1038/s41586-018-0500-9 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
