

Clinical analysis for 15 patients with pulmonary Langerhans cell histiocytosis and literature review
- PMID: 35545326
- PMCID: PMC10930052
- DOI: 10.11817/j.issn.1672-7347.2022.210581
Clinical analysis for 15 patients with pulmonary Langerhans cell histiocytosis and literature review
Abstract
Objectives: Pulmonary Langerhans cell histiocytosis (PLCH) is a clonal disease, characterized by proliferation of Langerhans cells that derived from bone marrow infiltrating the lungs and other organs. Due to the rarity of the disease, the current understanding of the disease is insufficient, often misdiagnosed or missed diagnosis. This study aims to raise clinicians' awareness for this disease via summarizing the clinical characteristics, imaging features, and treatment of PLCH.
Methods: We retrospectively analyzed clinical and follow-up data of 15 hospitalized cases of PLCH from September 2012 to June 2021 in the Second Xiangya Hospital of Central South University.
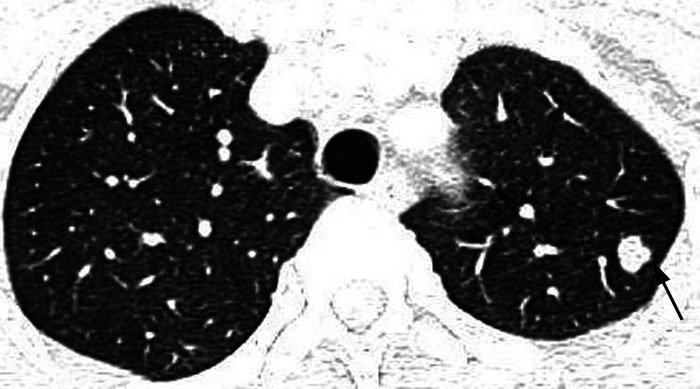
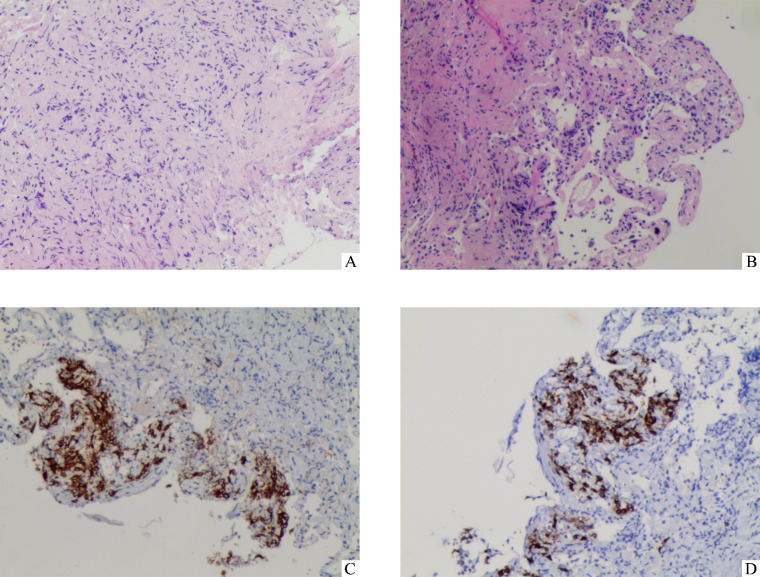
Results: The age of 15 patients (9 men and 6 women, with a sex ratio of 3 to 2) was 21-52 (median 33) years. Among them, 8 had a history of smoking and 5 suffered spontaneous pneumothorax during disease course. There were 3 patients with single system PLCH and 12 patients with multi-system PLCH, including 7 patients with pituitary involvement, 7 patients with lymph node involvement, 6 patients with bone involvement, 5 patients with liver involvement, 2 patients with skin involvement, 2 patients with thyroid involvement, and 1 patients with thymus involvement. The clinical manifestations were varied but non-specific. Respiratory symptoms mainly included dry cough, sputum expectoration, chest pain, etc. Constitutional symptoms included fever and weight loss. Patients with multi-system involvement experienced symptoms such as polyuria-polydipsia, bone pain, and skin rash. All patients were confirmed by pathology, including 6 by lung biopsy, 3 by bone biopsy, 2 by lymph node biopsy, and 4 by liver, skin, suprasternal fossa tumor, or pituitary stalk biopsy. The most common CT findings from this cohort of patients were nodules and/or cysts and nodular and cystic shadows were found in 7 patients. Three patients presented simple multiple cystic shadows, 3 patients presented multiple nodules, and 2 patients presented with single nodules and mass shadows. Pulmonary function tests were performed in 4 patients, ventilation dysfunction was showed in 2 patients at the first visit. Pulmonary diffusion function tests were performed in 4 patients and showed a decrease in 3 patients. Smoking cessation was recommended to PLCH patients with smoking history. Ten patients received chemotherapy while 2 patients received oral glucocorticoid therapy. Among the 11 patients with the long-term follow-up, 9 were in stable condition.
Conclusions: PLCH is a neoplastic disease closely related to smoking. The clinical manifestations and laboratory examination are not specific. Pneumothorax could be the first symptom which is very suggestive of the disease. Definitive diagnosis relies on histology. There is no unified treatment plan for PLCH, and individualized treatment should be carried out according to organ involvement. Early smoking cessation is essential. Chemotherapy is the main treatment for rapidly progressing PLCH involved multiple organs. All diagnosed patients can be considered for the detection of BRAFV600E gene and relevant targeted therapies have been implemented recently.
目的: 肺朗格汉斯细胞组织细胞增生症(pulmonary Langerhans cell histiocytosis,PLCH)是一种以朗格汉斯细胞增殖浸润肺及其他器官为主要病理特征的克隆性疾病。由于该病罕见,目前对该病的认识不足,经常被误诊或漏诊。本研究旨在通过总结PLCH的临床特征、影像学特征和治疗情况,提高临床医生对该病的认识。方法: 收集2012年9月至2021年6月中南大学湘雅二医院收治的15例PLCH患者的临床和随访资料并进行分析和总结。结果: 在15例PLCH患者中,男女比例为3꞉2,年龄21~52(中位年龄33)岁,其中8例患者有吸烟史。5例患者首次发病时或病程中出现自发性气胸。单系统PLCH 3例,多系统PLCH 12例。垂体受累7例,淋巴结受累7例,骨受累6例,肝受累5例,皮肤受累2例,甲状腺受累2例,胸腺受累1例。临床症状表现多样,无特异性。肺部症状常表现为干咳、咳痰、胸痛等;全身症状主要表现为消瘦、乏力、发热,多系统累及者可出现口干、多尿、骨痛、皮疹等症状。所有患者经病理确诊,经肺活检确诊6例,骨活检3例,淋巴结活检2例,肝、皮肤、胸骨上窝肿块、垂体柄活检各1例。肺部影像以结节影和/或囊状影为主要表现,7例表现为结节影、囊状影并存;3例呈单纯多发囊状影;3例呈多发结节影;2例表现为单发结节、肿块影。4例患者确诊时行肺功能检查,2例患者出现肺通气功能障碍;4例患者确诊时行肺弥散功能检查,其中3例出现弥散功能下降。有吸烟史的患者均行戒烟治疗。10例患者接受化学药物治疗,2例患者接受口服糖皮质激素治疗。11例患者长期随访,9例病情稳定。结论: PLCH是一种肿瘤性疾病,与吸烟密切相关。临床表现及实验室检查无特异性,气胸为首发症状时对诊断该病具有重要提示意义。确诊主要依靠病理检查。PLCH尚无统一治疗方案,应根据器官受累情况进行个体化治疗。早期戒烟对治疗至关重要,化学药物治疗为快速进展型及累及多器官PLCH的主要治疗手段。确诊的患者均可考虑行BRAFV600E基因检测,相关靶向治疗将成为未来治疗的热点。.
Keywords: clinical manifestation; imaging features; prognosis; pulmonary Langerhans cell histiocytosis; treatment.
Conflict of interest statement
作者声称无任何利益冲突。
Figures


Similar articles
-
Pulmonary Langerhans cell histiocytosis with cervical lymph node involvement, and coexistence with pulmonary tuberculosis and right pneumothorax: a case report and review of literature.Int J Clin Exp Pathol. 2015 Feb 1;8(2):2146-52. eCollection 2015. Int J Clin Exp Pathol. 2015. PMID: 25973117 Free PMC article. Review.
-
Pulmonary langerhans cell histiocytosis secondary to Marijuana use: a case report and systematic review of the literature.BMC Pulm Med. 2025 Jan 27;25(1):44. doi: 10.1186/s12890-025-03513-3. BMC Pulm Med. 2025. PMID: 39871311 Free PMC article.
-
Pulmonary Langerhans Cell Histiocytosis: An Update From the Pathologists' Perspective.Arch Pathol Lab Med. 2016 Mar;140(3):230-40. doi: 10.5858/arpa.2015-0246-RA. Arch Pathol Lab Med. 2016. PMID: 26927717 Review.
-
Pulmonary Langerhans cell histiocytosis: a comprehensive analysis of 40 patients and literature review.Eur J Intern Med. 2015 Jun;26(5):351-6. doi: 10.1016/j.ejim.2015.04.001. Epub 2015 Apr 17. Eur J Intern Med. 2015. PMID: 25899682 Review.
-
Pulmonary Langerhans cell histiocytosis; characteristics of 11 cases.Tuberk Toraks. 2013;61(4):333-41. doi: 10.5578/tt.5944. Tuberk Toraks. 2013. PMID: 24506750
Cited by
-
Is pulmonary Langerhans cell histiocytosis really rare?: A retrospective cohort study.Medicine (Baltimore). 2025 Aug 8;104(32):e43766. doi: 10.1097/MD.0000000000043766. Medicine (Baltimore). 2025. PMID: 40797453 Free PMC article.
-
Pulmonary Langerhans cell histiocytosis presenting as an uncommon mass in the lung: A rare case report and literature review.Clin Case Rep. 2024 Aug 19;12(8):e9342. doi: 10.1002/ccr3.9342. eCollection 2024 Aug. Clin Case Rep. 2024. PMID: 39161669 Free PMC article.
References
-
- Salama HA, Jazieh AR, Alhejazi AY, et al. . Highlights of the management of adult histiocytic disorders: Langerhans cell histiocytosis, erdheim-Chester disease, rosai-dorfman disease, and hemophagocytic lymphohistiocytosis[J/OL]. Clin Lymphoma Myeloma Leuk, 2021, 21(1): e66-e75 [2021-09-01]. 10.1016/j.clml.2020.08.007. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
