

Extricating human tumour immune alterations from tissue inflammation
- PMID: 35545675
- PMCID: PMC9132772
- DOI: 10.1038/s41586-022-04718-w
Extricating human tumour immune alterations from tissue inflammation
Abstract
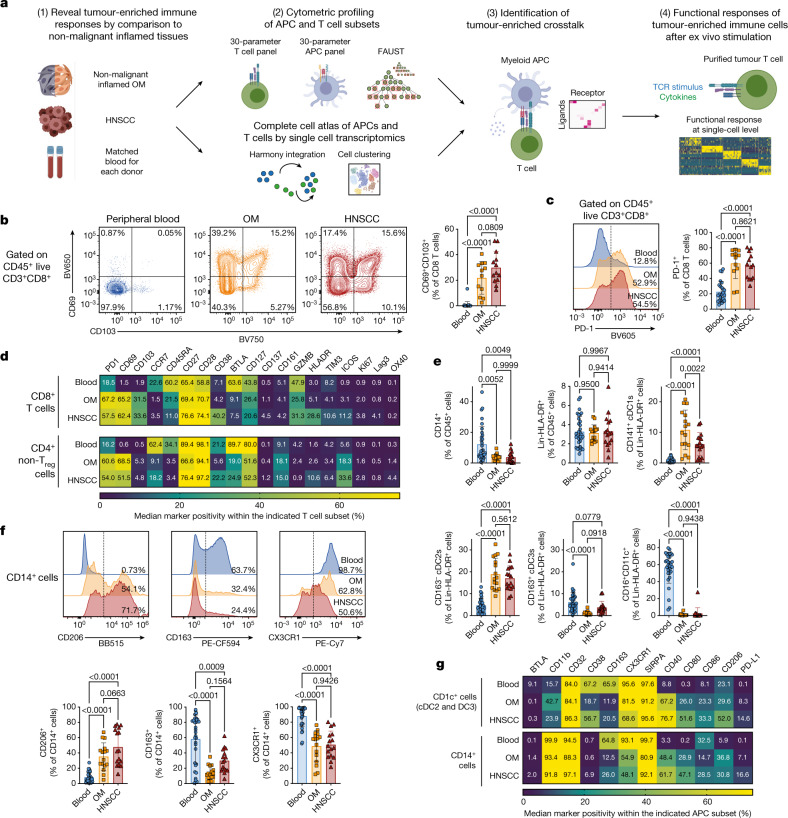
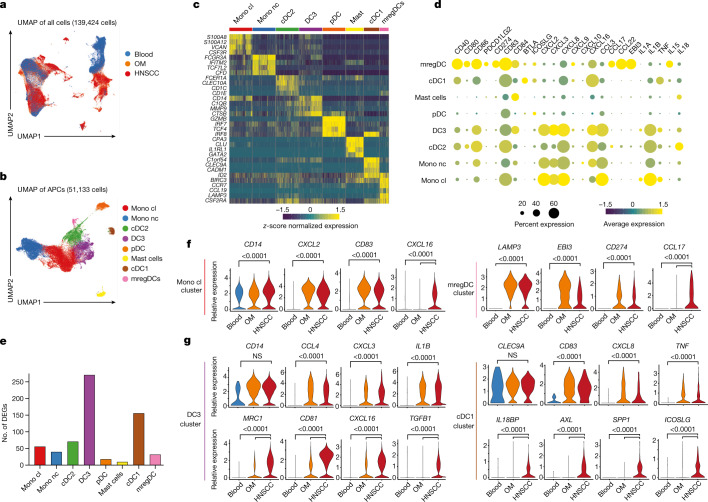
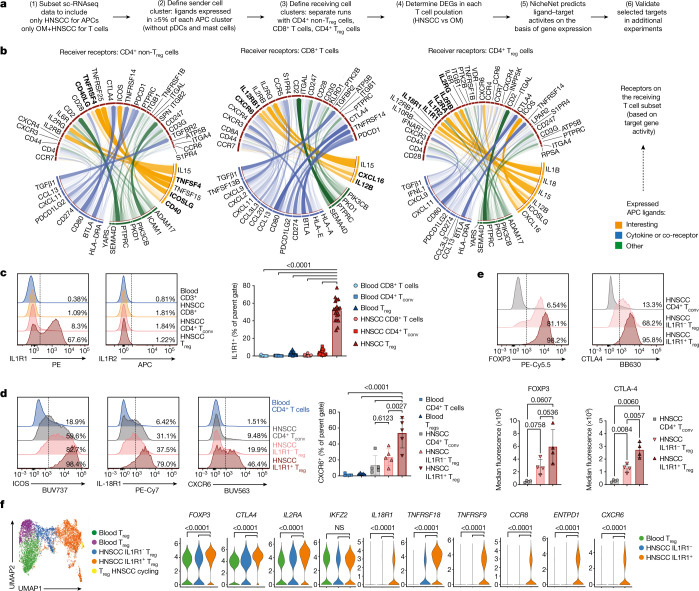
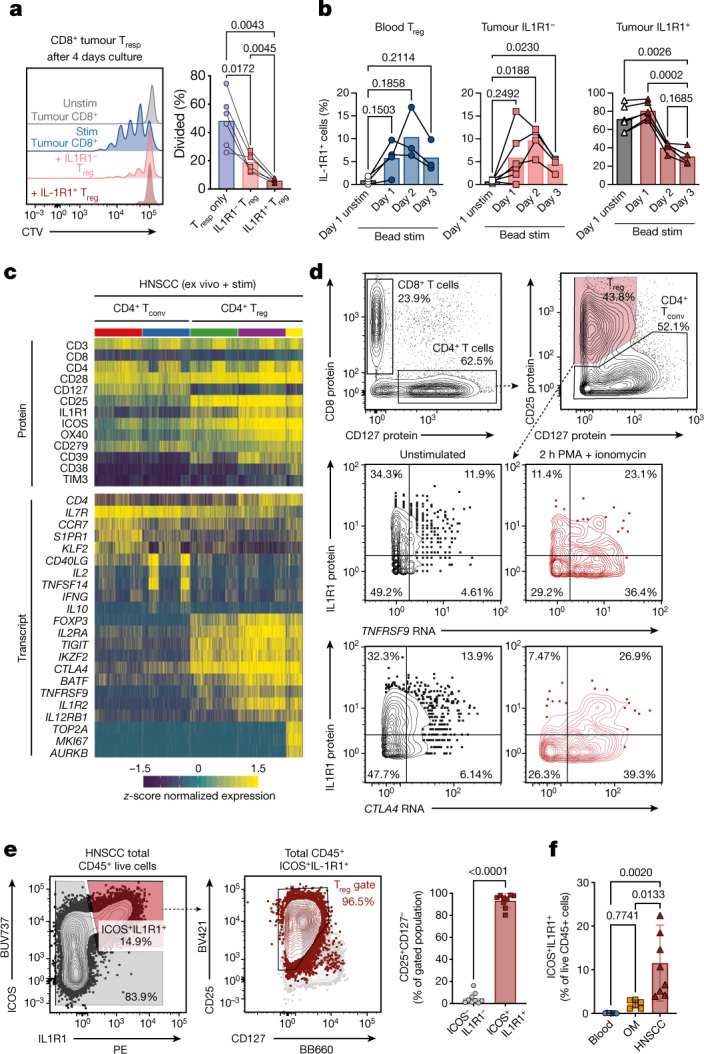
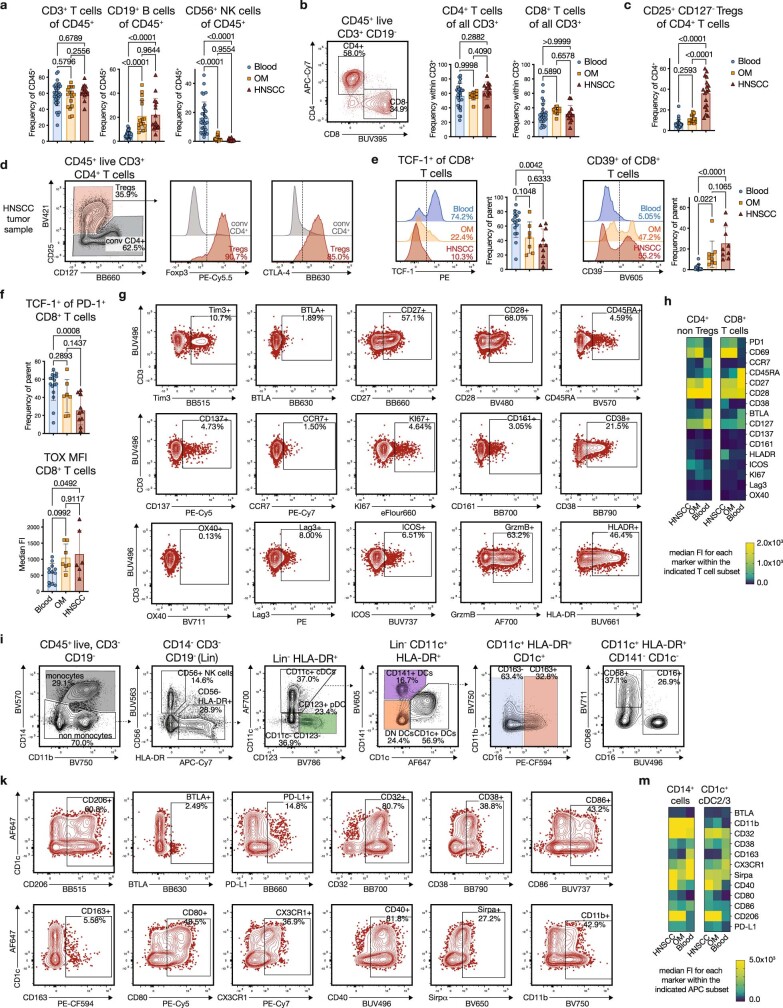
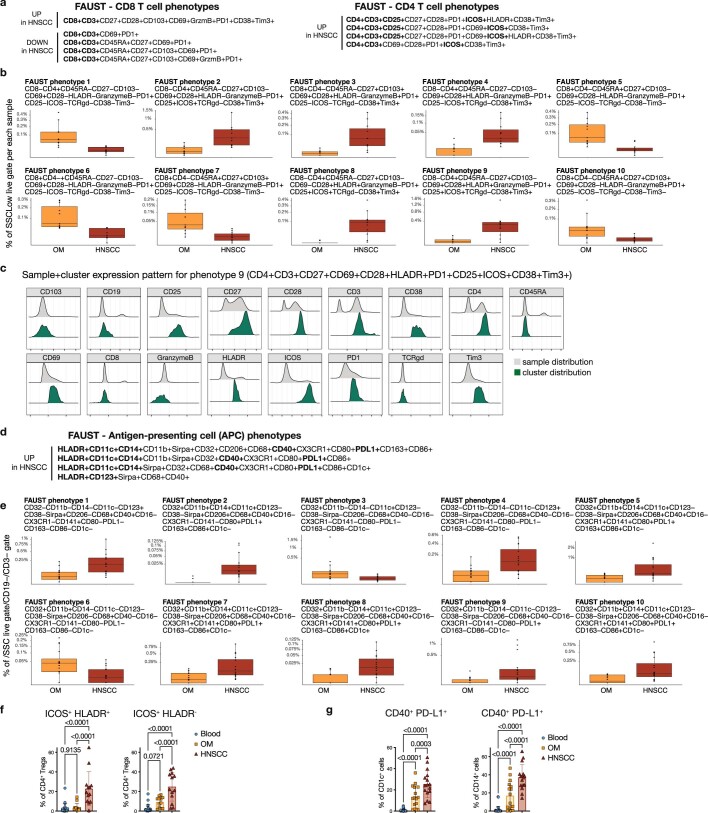
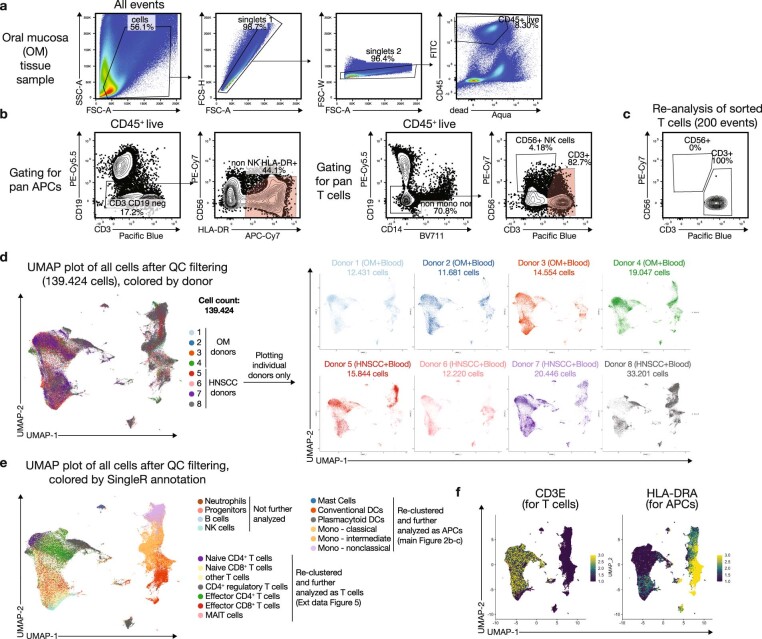
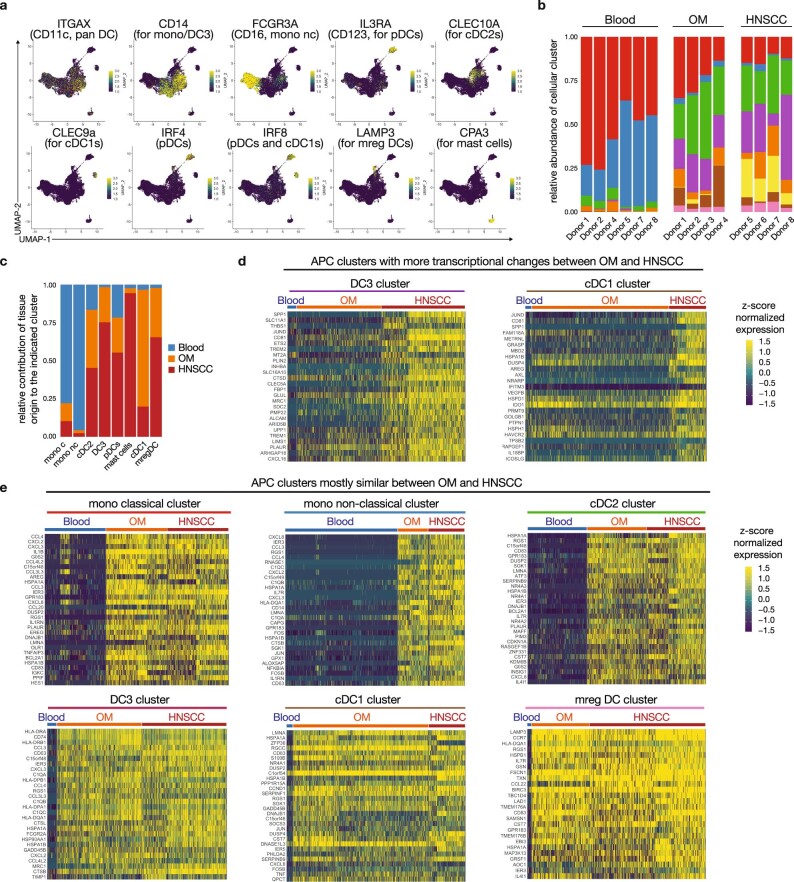
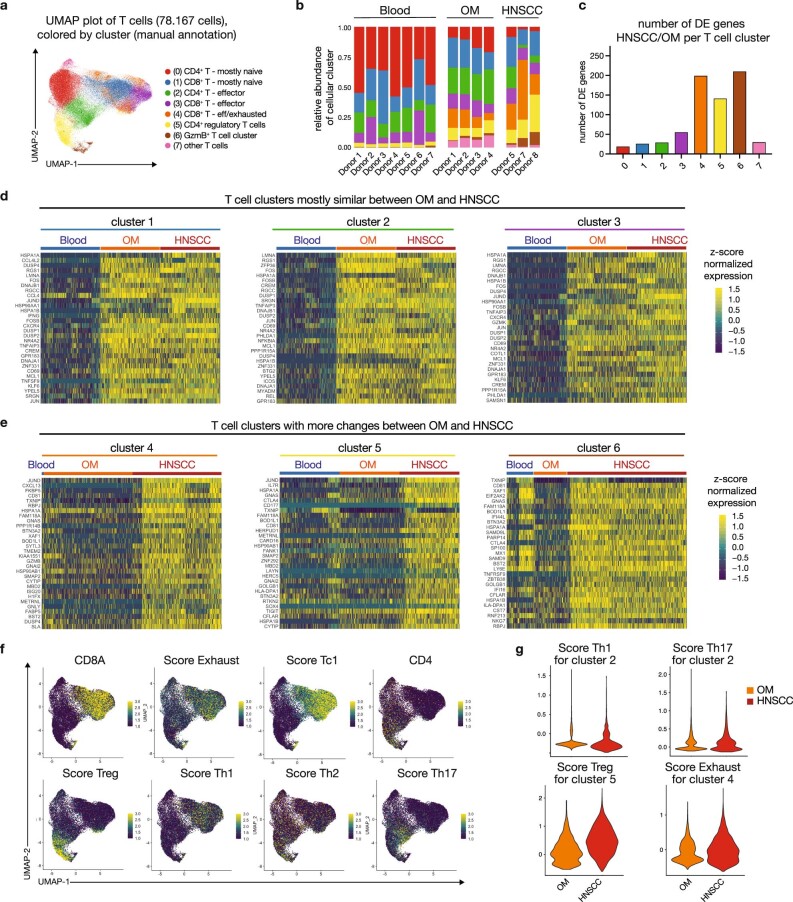
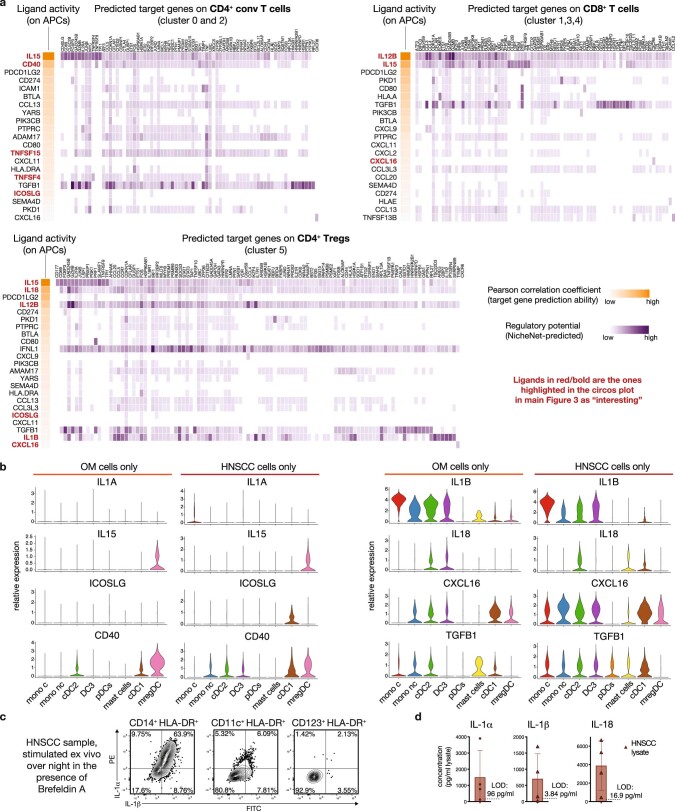
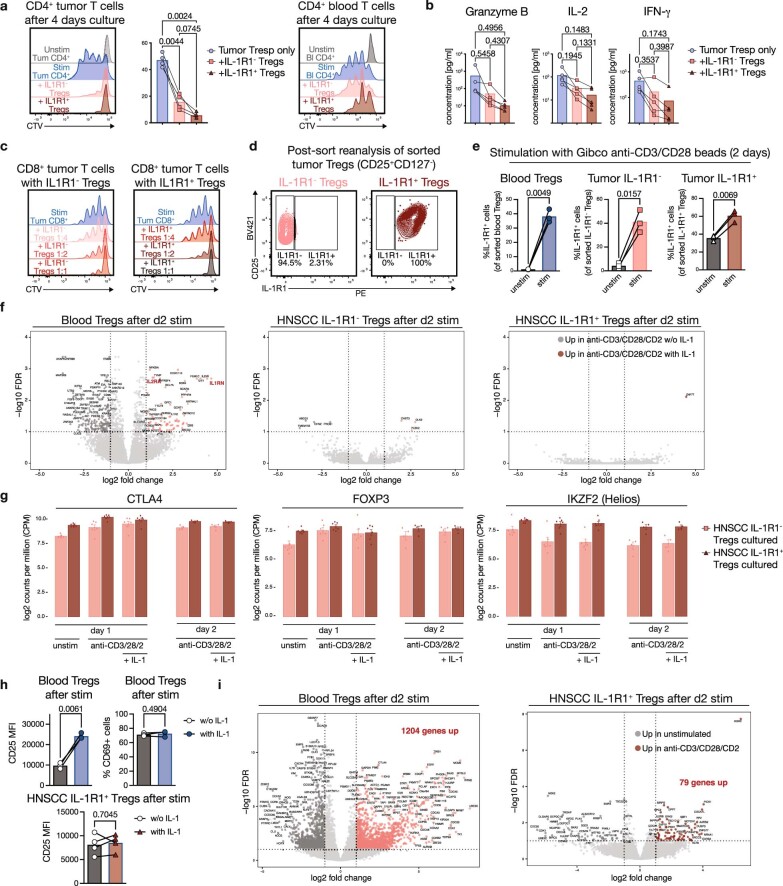
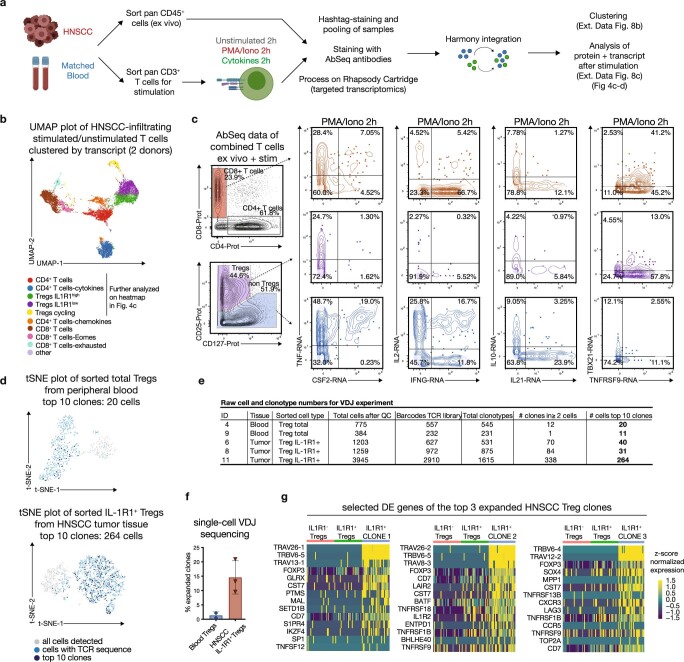
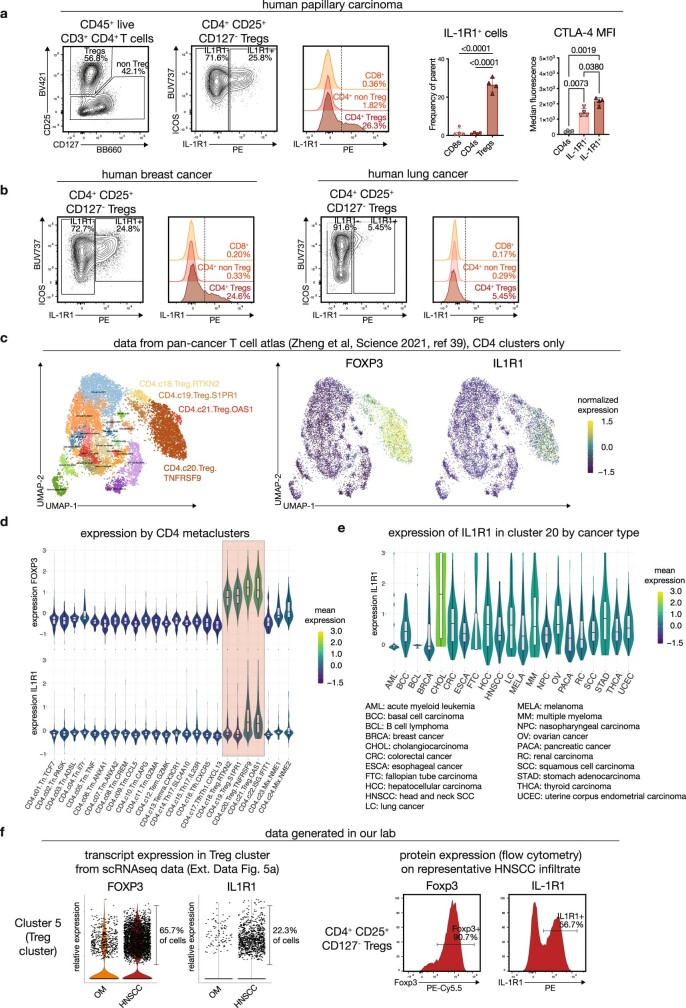
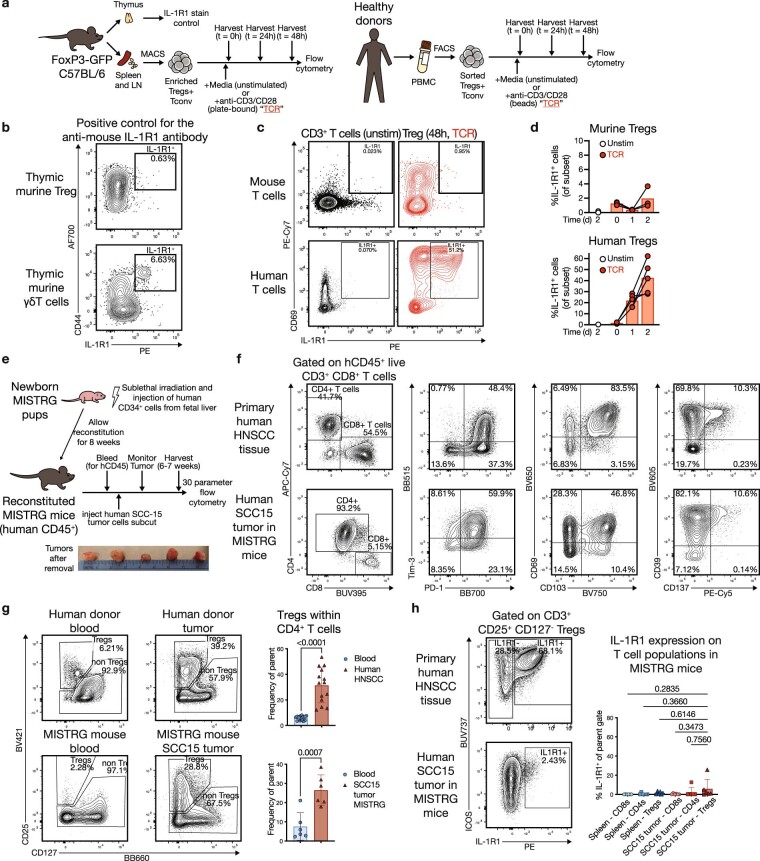
Immunotherapies have achieved remarkable successes in the treatment of cancer, but major challenges remain1,2. An inherent weakness of current treatment approaches is that therapeutically targeted pathways are not restricted to tumours, but are also found in other tissue microenvironments, complicating treatment3,4. Despite great efforts to define inflammatory processes in the tumour microenvironment, the understanding of tumour-unique immune alterations is limited by a knowledge gap regarding the immune cell populations in inflamed human tissues. Here, in an effort to identify such tumour-enriched immune alterations, we used complementary single-cell analysis approaches to interrogate the immune infiltrate in human head and neck squamous cell carcinomas and site-matched non-malignant, inflamed tissues. Our analysis revealed a large overlap in the composition and phenotype of immune cells in tumour and inflamed tissues. Computational analysis identified tumour-enriched immune cell interactions, one of which yields a large population of regulatory T (Treg) cells that is highly enriched in the tumour and uniquely identified among all haematopoietically-derived cells in blood and tissue by co-expression of ICOS and IL-1 receptor type 1 (IL1R1). We provide evidence that these intratumoural IL1R1+ Treg cells had responded to antigen recently and demonstrate that they are clonally expanded with superior suppressive function compared with IL1R1- Treg cells. In addition to identifying extensive immunological congruence between inflamed tissues and tumours as well as tumour-specific changes with direct disease relevance, our work also provides a blueprint for extricating disease-specific changes from general inflammation-associated patterns.
© 2022. The Author(s).
Conflict of interest statement
Part of the single-cell sequencing experiments was supported with reagents received from BD Biosciences and Illumina. BD Biosciences and Illumina were not involved in data analysis and interpretation. R.G. has received speaker fees from Illumina and Fluidigm and support from Juno Therapeutics and Janssen Pharma, has consulted for Takeda Vaccines, Juno Therapeutics and Infotech Soft, and has ownership interest in Modulus Therapeutics. E.G. and R.G. declare ownership interest in Ozette Technologies. F.M., J.R.E. and M.P. hold the related patent
Figures
Comment in
-
Deciphering immunoregulatory vulnerabilities in human cancers.Nat Immunol. 2022 Jul;23(7):995-996. doi: 10.1038/s41590-022-01251-w. Nat Immunol. 2022. PMID: 35726061 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
