

Fundamental immune-oncogenicity trade-offs define driver mutation fitness
- PMID: 35545680
- PMCID: PMC9159948
- DOI: 10.1038/s41586-022-04696-z
Fundamental immune-oncogenicity trade-offs define driver mutation fitness
Erratum in
-
Author Correction: Fundamental immune-oncogenicity trade-offs define driver mutation fitness.Nature. 2022 Jun;606(7914):E5. doi: 10.1038/s41586-022-04879-8. Nature. 2022. PMID: 35641605 Free PMC article. No abstract available.
Abstract
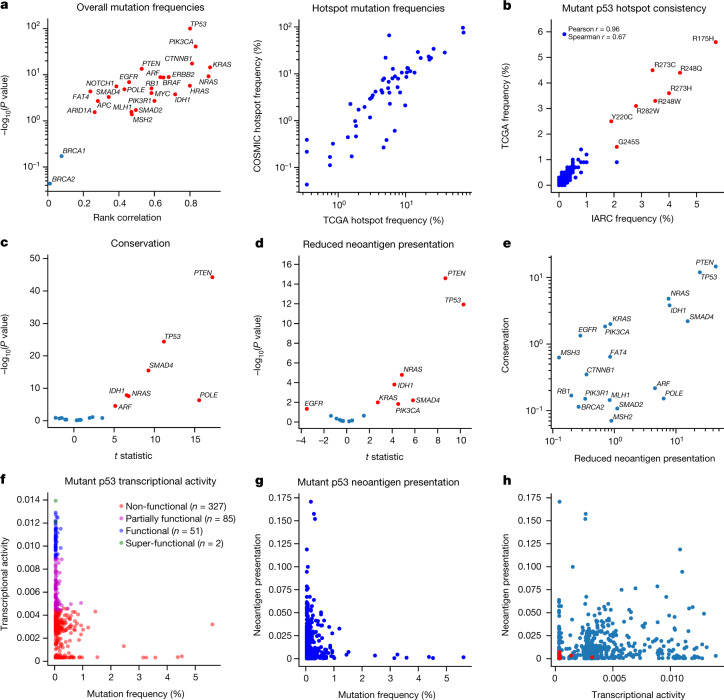
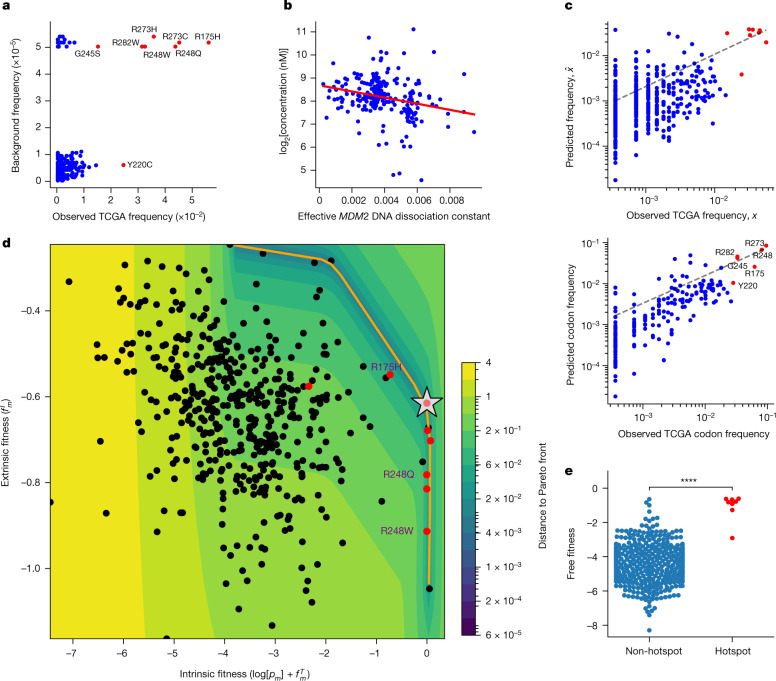
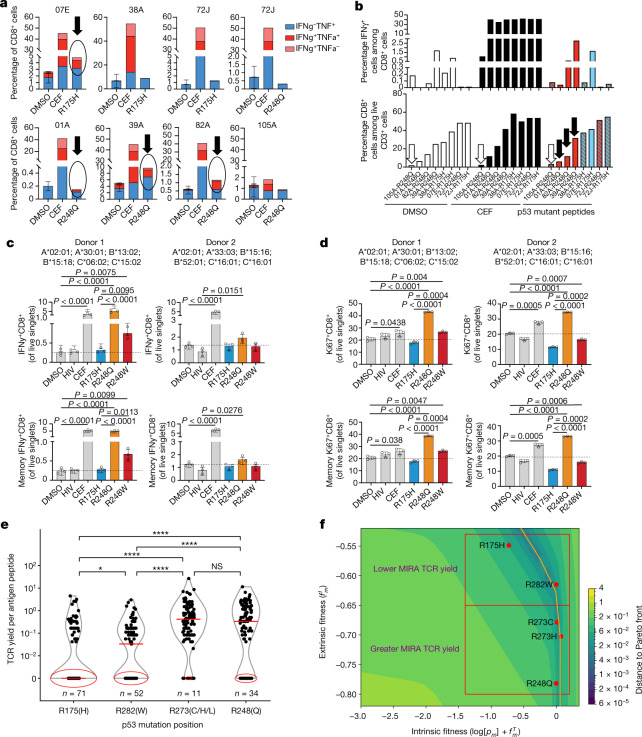
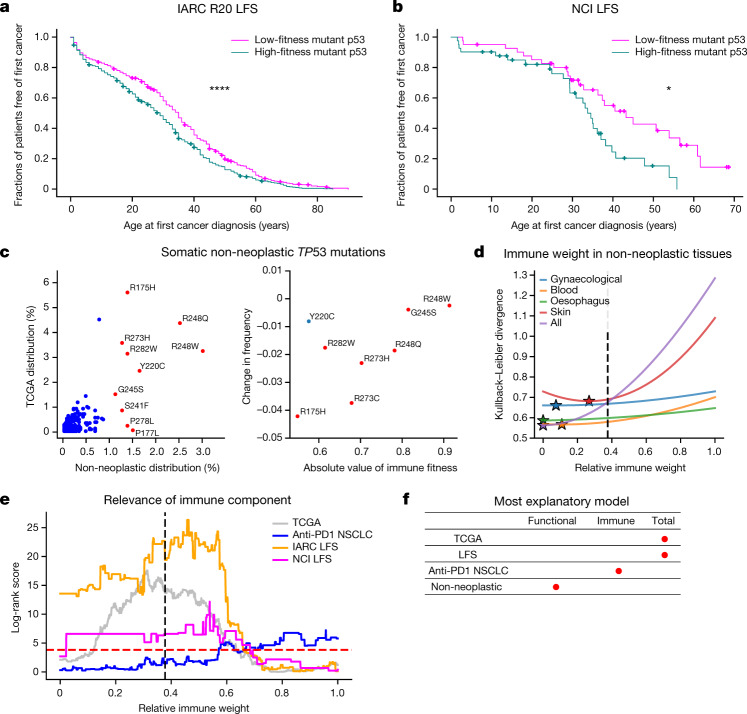
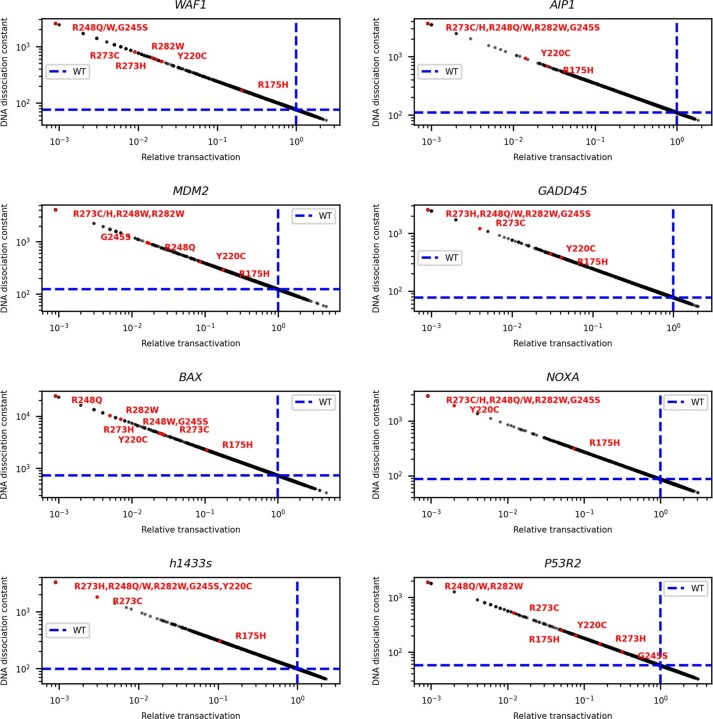
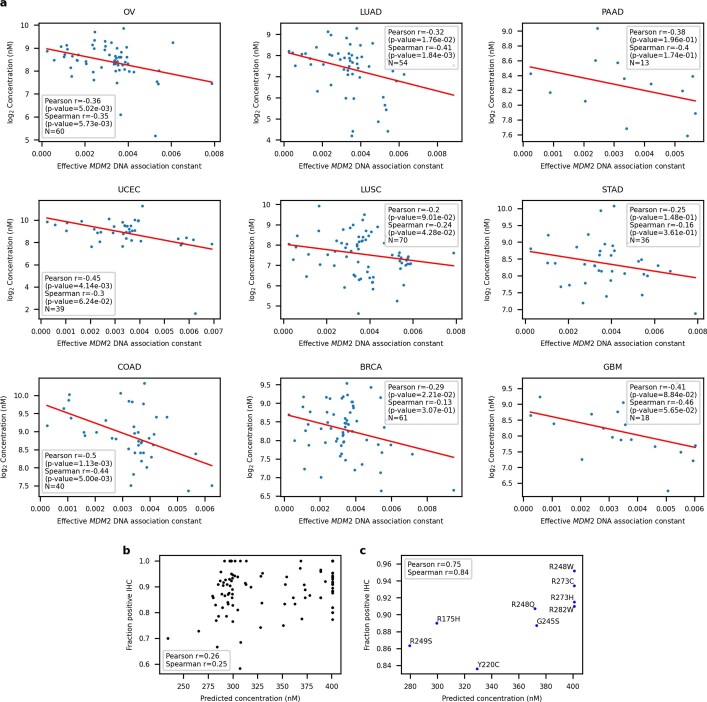
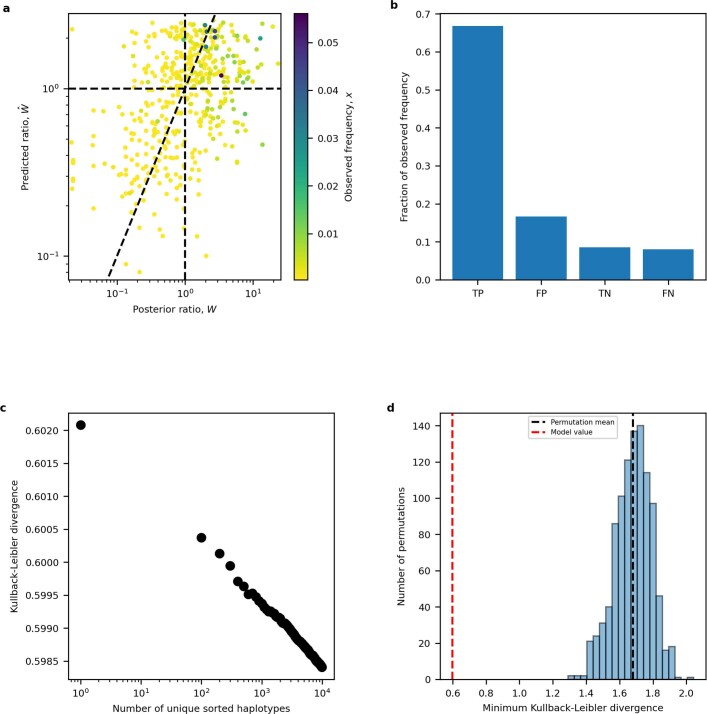
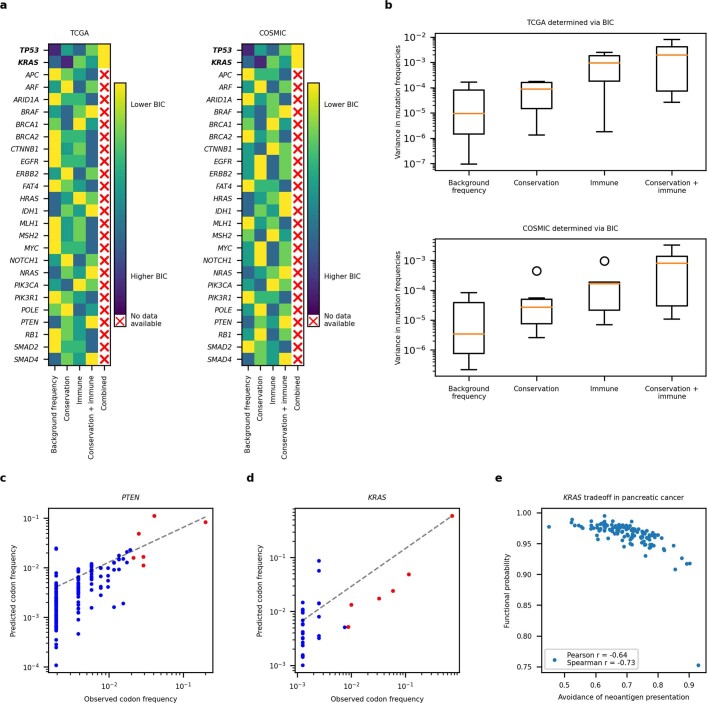
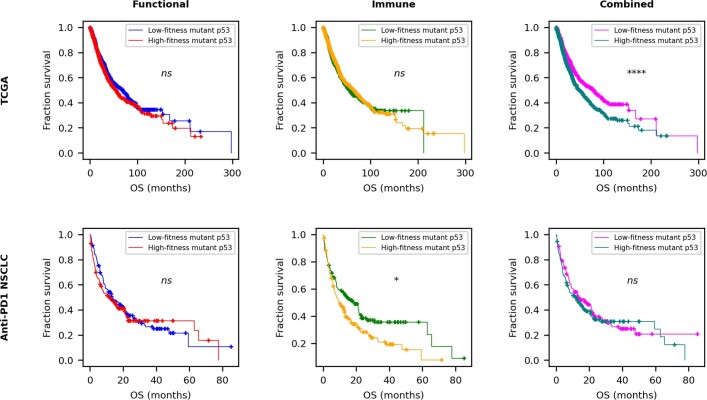
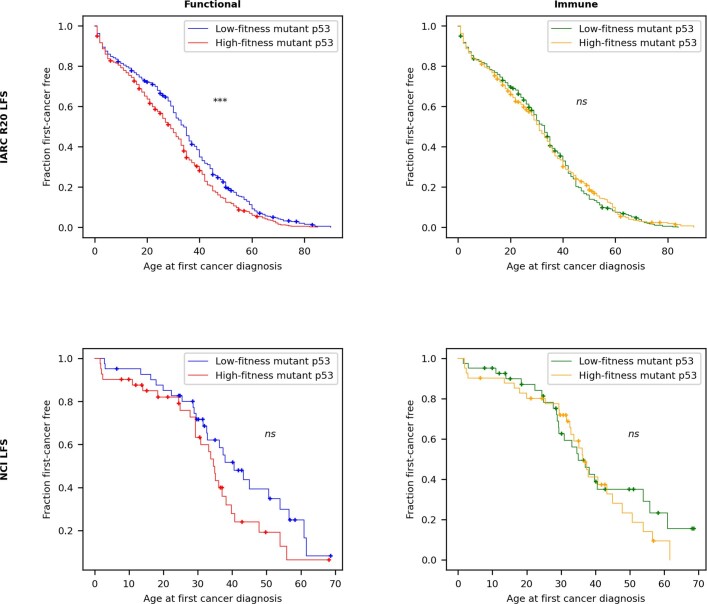
Missense driver mutations in cancer are concentrated in a few hotspots1. Various mechanisms have been proposed to explain this skew, including biased mutational processes2, phenotypic differences3-6 and immunoediting of neoantigens7,8; however, to our knowledge, no existing model weighs the relative contribution of these features to tumour evolution. We propose a unified theoretical 'free fitness' framework that parsimoniously integrates multimodal genomic, epigenetic, transcriptomic and proteomic data into a biophysical model of the rate-limiting processes underlying the fitness advantage conferred on cancer cells by driver gene mutations. Focusing on TP53, the most mutated gene in cancer1, we present an inference of mutant p53 concentration and demonstrate that TP53 hotspot mutations optimally solve an evolutionary trade-off between oncogenic potential and neoantigen immunogenicity. Our model anticipates patient survival in The Cancer Genome Atlas and patients with lung cancer treated with immunotherapy as well as the age of tumour onset in germline carriers of TP53 variants. The predicted differential immunogenicity between hotspot mutations was validated experimentally in patients with cancer and in a unique large dataset of healthy individuals. Our data indicate that immune selective pressure on TP53 mutations has a smaller role in non-cancerous lesions than in tumours, suggesting that targeted immunotherapy may offer an early prophylactic opportunity for the former. Determining the relative contribution of immunogenicity and oncogenic function to the selective advantage of hotspot mutations thus has important implications for both precision immunotherapies and our understanding of tumour evolution.
© 2022. The Author(s).
Conflict of interest statement
D.F.B. is a consultant for Bristol Myers Squibb, Merck, Genentech–Roche, AstraZeneca and Pfizer and has received research support from Merck, Genentech–Roche, AstraZeneca, Novartis and Bristol Myers Squibb. M.K.C. has received consulting fees for Bristol Meyers Squibb, Merck, Incyte, Moderna, Immunocore and AstraZeneca and research funding from Bristol Meyers Squibb. L.F.C., E.J.O., M.K. and H.S.R. are employees of Adaptive Biotechnologies. S.A.F. has received research support from AstraZeneca and Genentech–Roche; is a consultant and advisory board member for Merck; and owns stock in UroGen, Allogene Therapeutics, Neogene Therapeutics, Kronos Bio and IconOVir. B.D.G. has received honoraria for speaking engagements from Merck, Bristol Meyers Squibb and Chugai Pharmaceutical; has received research funding from Bristol Meyers Squibb; has been a compensated consultant for PMV Pharma, DarwinHealth and ROME Therapeutics; and is a cofounder of ROME Therapeutics. M.D.H. reports personal fees from Achilles, Adagene, Adicet, Arcus, AstraZeneca, Blueprint, Bristol Myers Squib, Da Volterra, Eli Lilly, Genentech–Roche, Genzyme–Sanofi, Janssen, Immunai, Instil Bio, Mana Therapeutics, Merck, Mirati, Natera, PACT Pharma, Shattuck Labs and Regeneron and has equity options with Factorial, Immunai, Shattuck Labs and Arcus. M.D.H. also reports that a patent filed by Memorial Sloan Kettering related to the use of tumour mutational burden to predict response to immunotherapy (PCT/US2015/062208) is pending and licensed by Personal Genome Diagnostics and that, subsequent to completing this work, he became an employee of AstraZeneca. A.J.L. is a founder, director and shareholder of PMV Pharma and is the chair of the Janssen scientific advisory board. T.M. is a cofounder and holds equity in Imvaq Therapeutics; is a consultant for ImmunOs Therapeutics, Im AQ19 munoGenesis and Pfizer; has received research support from Bristol Myers Squibb, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals, Peregrine Pharmaceuticals, Adaptive Biotechnologies, Leap Therapeutics and Aprea; and has patent applications related to work on oncolytic viral therapy, alpha virus-based vaccines, neoantigen modelling, CD40, GITR, OX40, PD-1 and CTLA-4. I.S. is an inventor on a patent application related to work on CD40. J.E.R. has received consulting fees and trial funding from Bayer, Seagen, AstraZeneca, Roche, Astellas Pharma and QED Therapeutics; consulting fees from Bristol Myers Squibb, Merck, Pfizer, Pharmacyclics, Boehringer Ingelheim, GlaxoSmithKline, Infinity, Janssen, Mirati, EMD Serono, Gilead, BioClin, Eli Lilly and Company, Tyra Biosciences and Pharmacyclics; honoraria for continuing medical education from Research to Practice, MJH Life Sciences, Medscape, Clinical Care Options, OncLive and EMD Serono; royalties from UpToDate. B.W. reports ad hoc membership of the scientific advisory board of Repare Therapeutics, outside the submitted work. J.D.W. is a consultant for Adaptive Biotechnologies, Amgen, Apricity, Ascentage Pharma, ArsenalBio, Astellas, AstraZeneca, Bayer, BeiGene, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Chugai, Eli Lilly, Elucida, F-Star, Georgiamune, Imvaq, Kyowa Kirin, Linneaus, Merck, Neon Therapeutics, Polynoma, PsiOxus, Recepta, Takara Bio, Trieza, Truvax, SELLAS, Serametrix, Surface Oncology, Syndax, Syntalogic and Werewolf Therapeutics; receives grant and research support from Bristol Myers Squibb and Sephora; and has equity in Tizona Pharmaceuticals, Adaptive Biotechnologies, Imvaq, BeiGene, Linneaus, Apricity, ArsenalBio and Georgiamune. R.Z. is an inventor on patent applications related to work on GITR, PD-1 and CTLA-4; is a scientific advisory board member of iTEOS Therapeutics; has consulted for Leap Therapeutics; and receives grant support from AstraZeneca and Bristol Myers Squibb. D.Z. is a consultant for Merck, Agenus, Hookipa Biotech, AstraZeneca, Western Oncolytics, Synthekine, MANA Therapeutics, Xencor, Memgen and Takeda; receives grant and research support from AstraZeneca, Roche and Plexxikon; holds stock options with ImmunOs Therapeutics, Calidi Biotherapeutics and Accurius; and has a patent related to use of Newcastle disease virus for cancer therapy with royalties paid by Merck.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
