

Degron masking outlines degronons, co-degrading functional modules in the proteome
- PMID: 35545699
- PMCID: PMC9095673
- DOI: 10.1038/s42003-022-03391-z
Degron masking outlines degronons, co-degrading functional modules in the proteome
Abstract
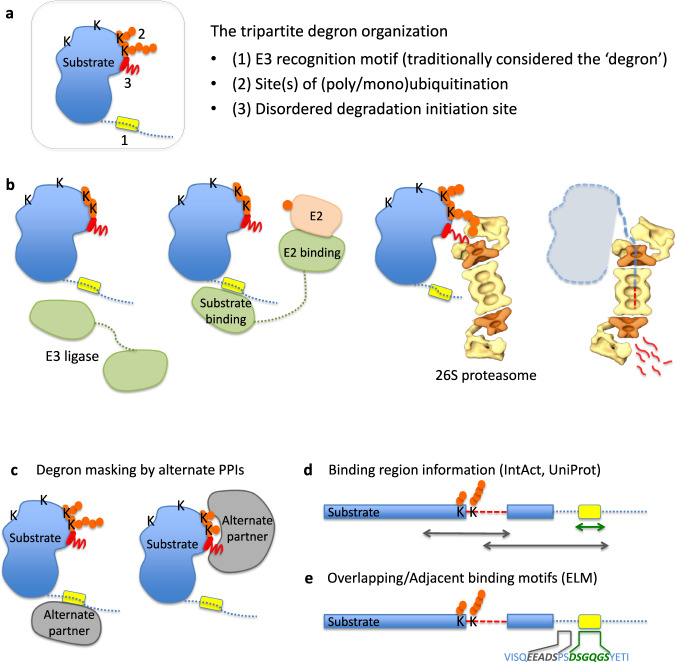
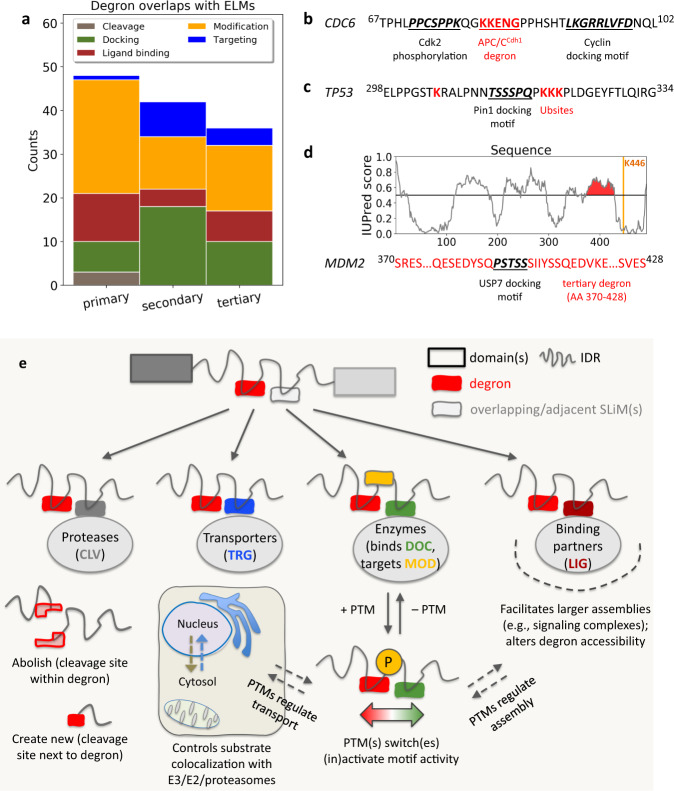
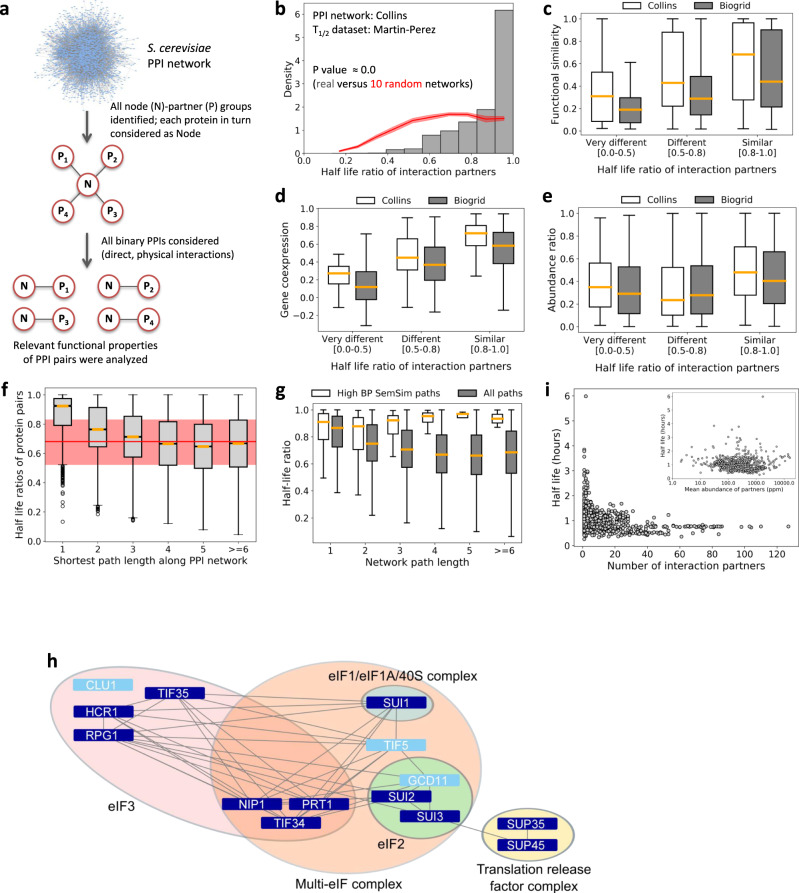
Effective organization of proteins into functional modules (networks, pathways) requires systems-level coordination between transcription, translation and degradation. Whereas the cooperation between transcription and translation was extensively studied, the cooperative degradation regulation of protein complexes and pathways has not been systematically assessed. Here we comprehensively analyzed degron masking, a major mechanism by which cellular systems coordinate degron recognition and protein degradation. For over 200 substrates with characterized degrons (E3 ligase targeting motifs, ubiquitination sites and disordered proteasomal entry sequences), we demonstrate that degrons extensively overlap with protein-protein interaction sites. Analysis of binding site information and protein abundance comparisons show that regulatory partners effectively outcompete E3 ligases, masking degrons from the ubiquitination machinery. Protein abundance variations between normal and cancer cells highlight the dynamics of degron masking components. Finally, integrative analysis of gene co-expression, half-life correlations and functional relationships between interacting proteins point towards higher-order, co-regulated degradation modules ('degronons') in the proteome.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
Systematic prediction of degrons and E3 ubiquitin ligase binding via deep learning.BMC Biol. 2022 Jul 14;20(1):162. doi: 10.1186/s12915-022-01364-6. BMC Biol. 2022. PMID: 35836176 Free PMC article.
-
The Eukaryotic Proteome Is Shaped by E3 Ubiquitin Ligases Targeting C-Terminal Degrons.Cell. 2018 Jun 14;173(7):1622-1635.e14. doi: 10.1016/j.cell.2018.04.028. Epub 2018 May 17. Cell. 2018. PMID: 29779948 Free PMC article.
-
A comparative analysis of the ubiquitination kinetics of multiple degrons to identify an ideal targeting sequence for a proteasome reporter.PLoS One. 2013 Oct 29;8(10):e78082. doi: 10.1371/journal.pone.0078082. eCollection 2013. PLoS One. 2013. PMID: 24205101 Free PMC article.
-
Design Principles Involving Protein Disorder Facilitate Specific Substrate Selection and Degradation by the Ubiquitin-Proteasome System.J Biol Chem. 2016 Mar 25;291(13):6723-31. doi: 10.1074/jbc.R115.692665. Epub 2016 Feb 5. J Biol Chem. 2016. PMID: 26851277 Free PMC article. Review.
-
Designing the Proteome with Chemical Tools: Degrons and Beyond.Chembiochem. 2025 Aug 22;26(15):e202500345. doi: 10.1002/cbic.202500345. Epub 2025 Jul 21. Chembiochem. 2025. PMID: 40399232 Review.
Cited by
-
ELM-the Eukaryotic Linear Motif resource-2024 update.Nucleic Acids Res. 2024 Jan 5;52(D1):D442-D455. doi: 10.1093/nar/gkad1058. Nucleic Acids Res. 2024. PMID: 37962385 Free PMC article.
-
Disordered clock protein interactions and charge blocks turn an hourglass into a persistent circadian oscillator.Nat Commun. 2024 Apr 25;15(1):3523. doi: 10.1038/s41467-024-47761-z. Nat Commun. 2024. PMID: 38664421 Free PMC article.
-
Degradation bottlenecks and resource competition in transiently and stably engineered mammalian cells.Nat Commun. 2025 Jan 2;16(1):328. doi: 10.1038/s41467-024-55311-w. Nat Commun. 2025. PMID: 39746977 Free PMC article.
-
N-SREBP2 Provides a Mechanism for Dynamic Control of Cellular Cholesterol Homeostasis.Cells. 2024 Jul 25;13(15):1255. doi: 10.3390/cells13151255. Cells. 2024. PMID: 39120286 Free PMC article.
-
Hierarchical graph learning for protein-protein interaction.Nat Commun. 2023 Feb 25;14(1):1093. doi: 10.1038/s41467-023-36736-1. Nat Commun. 2023. PMID: 36841846 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
