

Physicochemical Heuristics for Identifying High Fidelity, Near-Native Structural Models of Peptide/MHC Complexes
- PMID: 35547730
- PMCID: PMC9084917
- DOI: 10.3389/fimmu.2022.887759
Physicochemical Heuristics for Identifying High Fidelity, Near-Native Structural Models of Peptide/MHC Complexes
Abstract
There is long-standing interest in accurately modeling the structural features of peptides bound and presented by class I MHC proteins. This interest has grown with the advent of rapid genome sequencing and the prospect of personalized, peptide-based cancer vaccines, as well as the development of molecular and cellular therapeutics based on T cell receptor recognition of peptide-MHC. However, while the speed and accessibility of peptide-MHC modeling has improved substantially over the years, improvements in accuracy have been modest. Accuracy is crucial in peptide-MHC modeling, as T cell receptors are highly sensitive to peptide conformation and capturing fine details is therefore necessary for useful models. Studying nonameric peptides presented by the common class I MHC protein HLA-A*02:01, here we addressed a key question common to modern modeling efforts: from a set of models (or decoys) generated through conformational sampling, which is best? We found that the common strategy of decoy selection by lowest energy can lead to substantial errors in predicted structures. We therefore adopted a data-driven approach and trained functions capable of predicting near native decoys with exceptionally high accuracy. Although our implementation is limited to nonamer/HLA-A*02:01 complexes, our results serve as an important proof of concept from which improvements can be made and, given the significance of HLA-A*02:01 and its preference for nonameric peptides, should have immediate utility in select immunotherapeutic and other efforts for which structural information would be advantageous.
Keywords: major histocompatibility complex; neoantigen; peptide; prediction; structure; support vector machine.
Copyright © 2022 Keller, Weiss and Baker.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
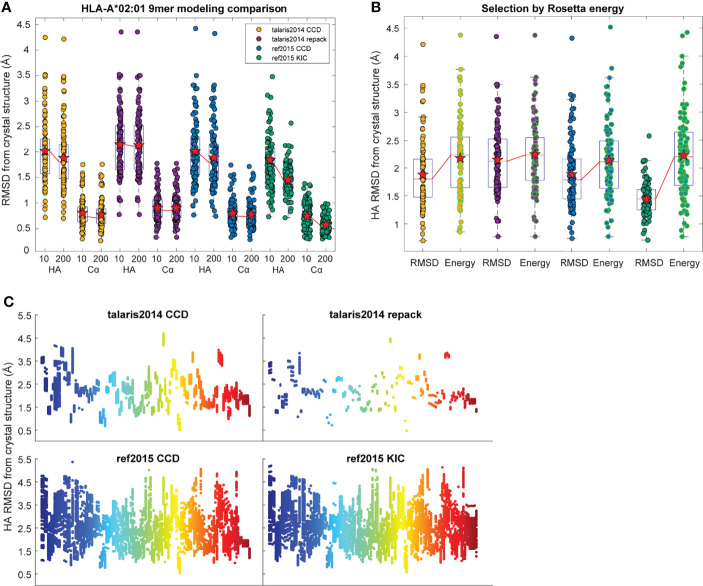
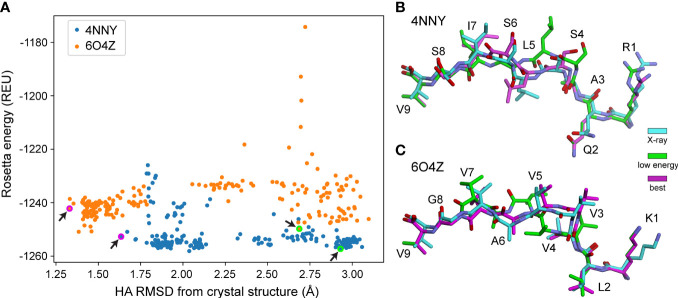



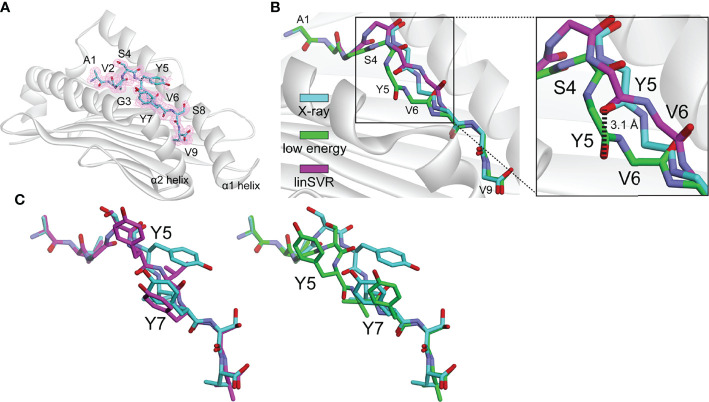

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials