

Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function
- PMID: 35549429
- PMCID: PMC9383269
- DOI: 10.1126/science.abl4290
Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function
Abstract
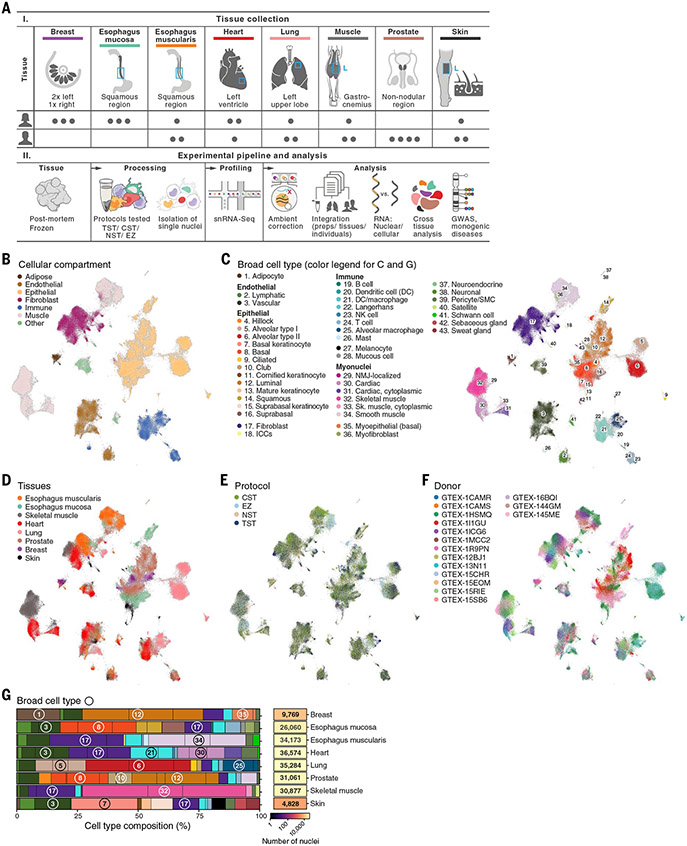
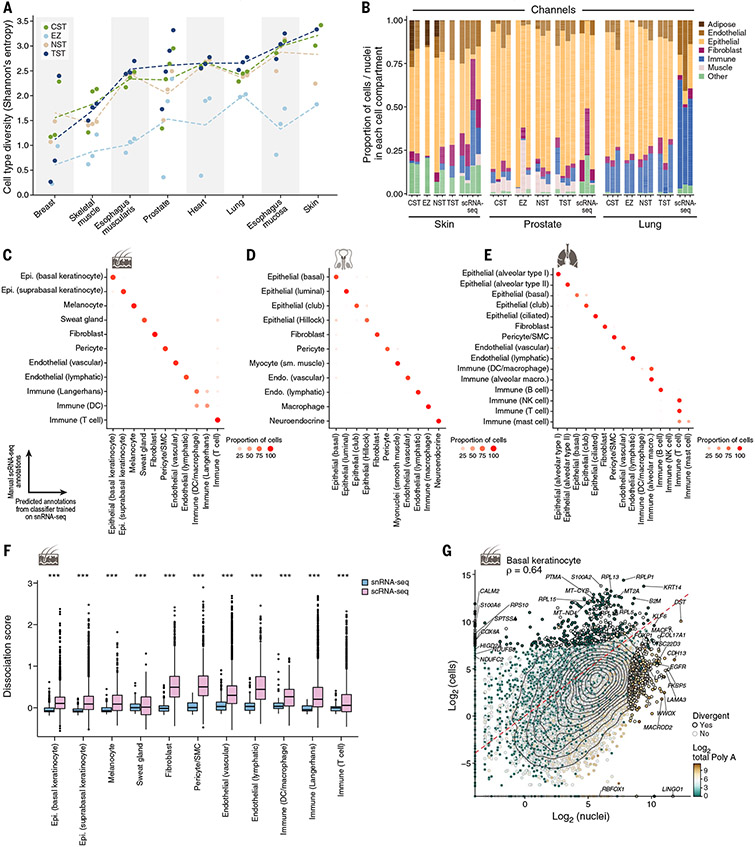
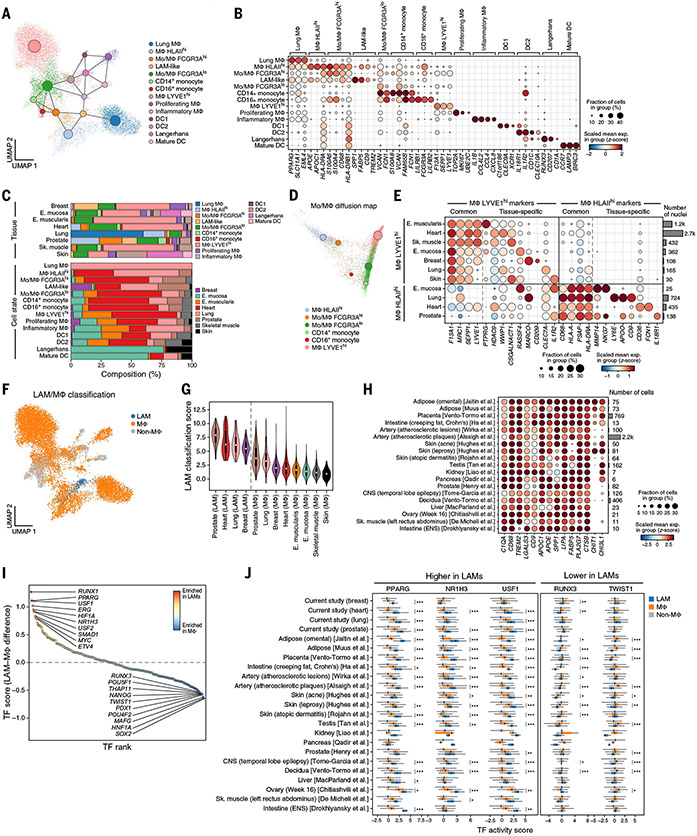
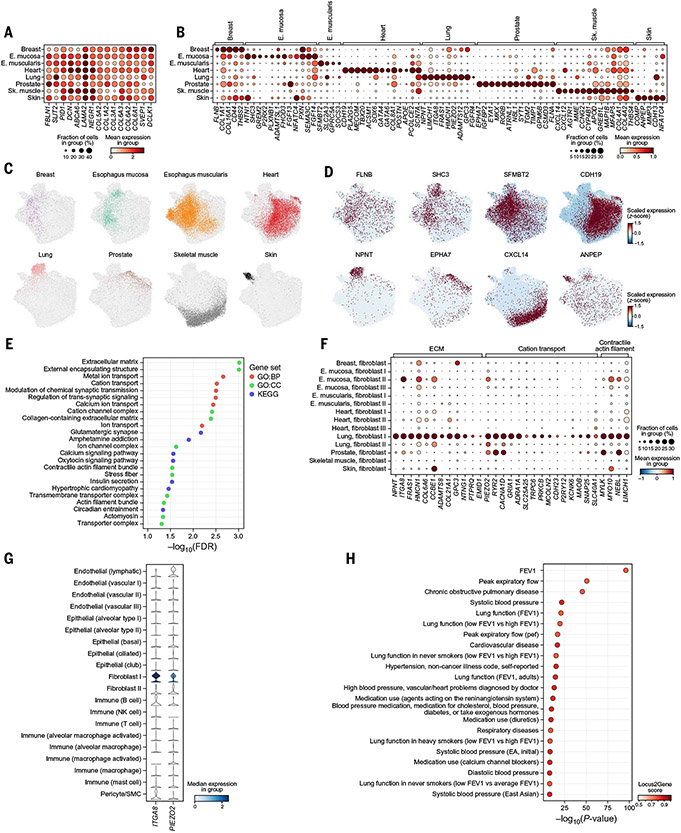
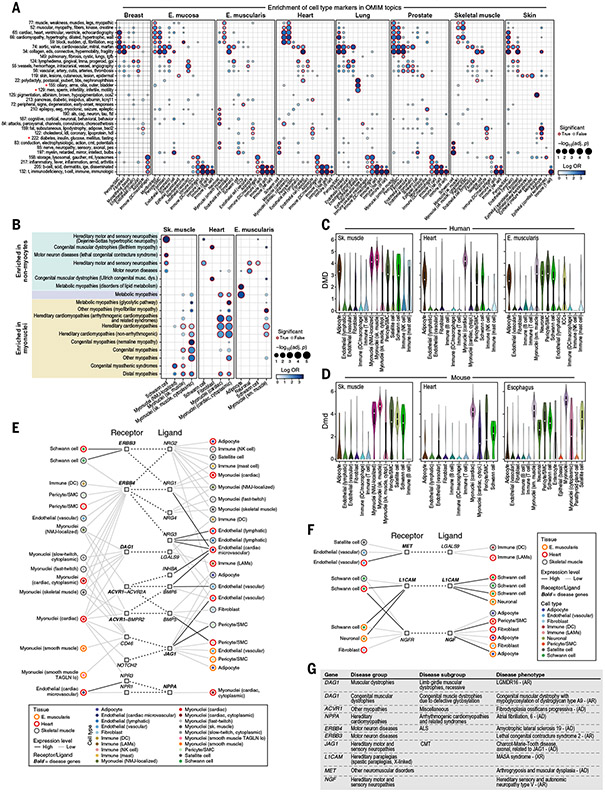
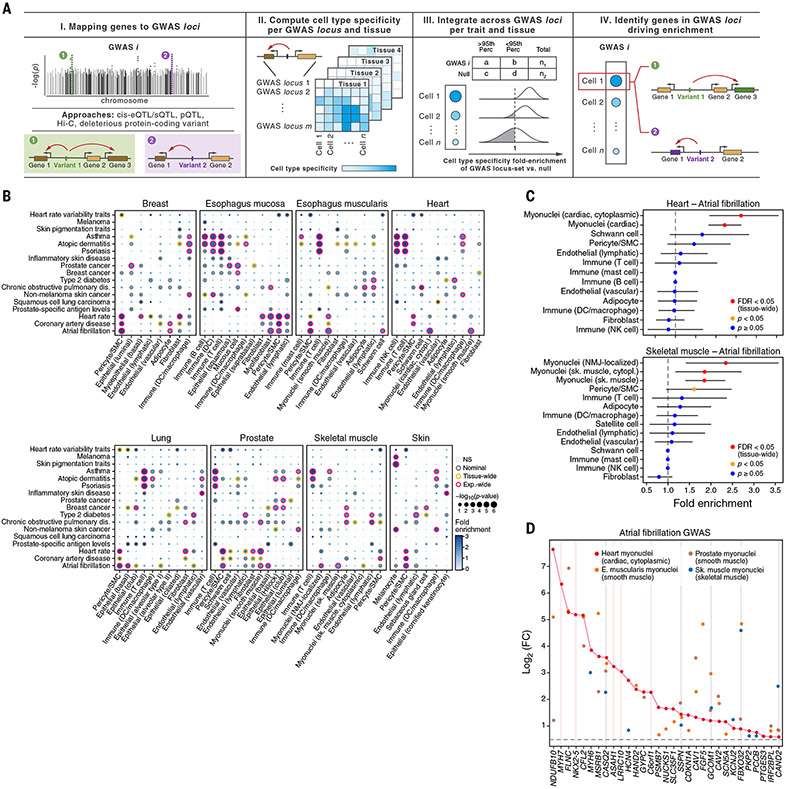
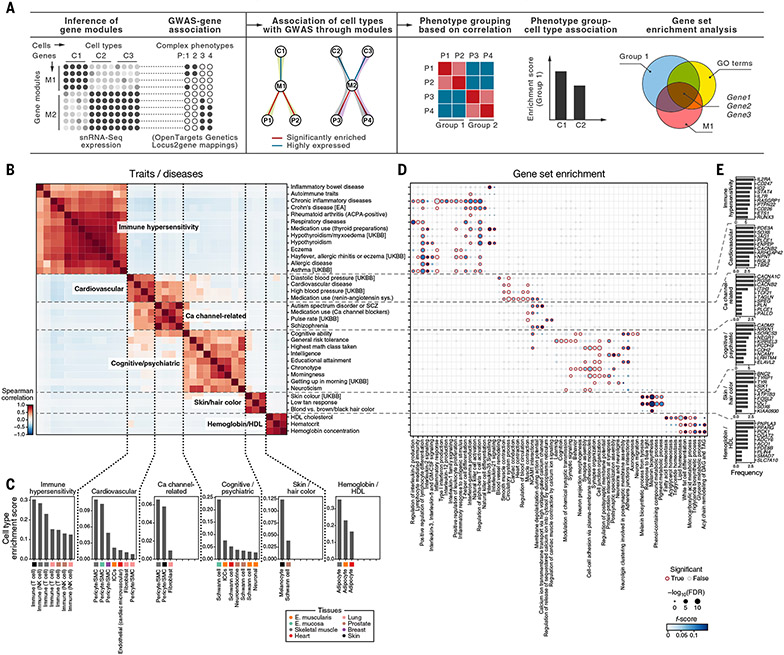
Understanding gene function and regulation in homeostasis and disease requires knowledge of the cellular and tissue contexts in which genes are expressed. Here, we applied four single-nucleus RNA sequencing methods to eight diverse, archived, frozen tissue types from 16 donors and 25 samples, generating a cross-tissue atlas of 209,126 nuclei profiles, which we integrated across tissues, donors, and laboratory methods with a conditional variational autoencoder. Using the resulting cross-tissue atlas, we highlight shared and tissue-specific features of tissue-resident cell populations; identify cell types that might contribute to neuromuscular, metabolic, and immune components of monogenic diseases and the biological processes involved in their pathology; and determine cell types and gene modules that might underlie disease mechanisms for complex traits analyzed by genome-wide association studies.
Figures
Comment in
-
Mapping cell types across human tissues.Science. 2022 May 13;376(6594):695-696. doi: 10.1126/science.abq2116. Epub 2022 May 12. Science. 2022. PMID: 35549410
-
Non-transplantable organs and tissues: A golden opportunity.Am J Transplant. 2022 Sep;22(9):2127-2128. doi: 10.1111/ajt.16671. Am J Transplant. 2022. PMID: 36039543 No abstract available.
-
Human cross-tissue cell atlases: unprecedented resources towards systematic understanding of physiology and diseases.Signal Transduct Target Ther. 2022 Oct 5;7(1):352. doi: 10.1038/s41392-022-01201-w. Signal Transduct Target Ther. 2022. PMID: 36198687 Free PMC article. No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
