

Taxonomic position, antibiotic resistance and virulence factor production by Stenotrophomonas isolates from patients with cystic fibrosis and other chronic respiratory infections
- PMID: 35549675
- PMCID: PMC9097388
- DOI: 10.1186/s12866-022-02466-5
Taxonomic position, antibiotic resistance and virulence factor production by Stenotrophomonas isolates from patients with cystic fibrosis and other chronic respiratory infections
Abstract
Background: The potential pathogenic role of Stenotrophomonas maltophilia in lung disease and in particular in cystic fibrosis is unclear. To develop further understanding of the biology of this taxa, the taxonomic position, antibiotic resistance and virulence factors of S. maltophilia isolates from patients with chronic lung disease were studied.
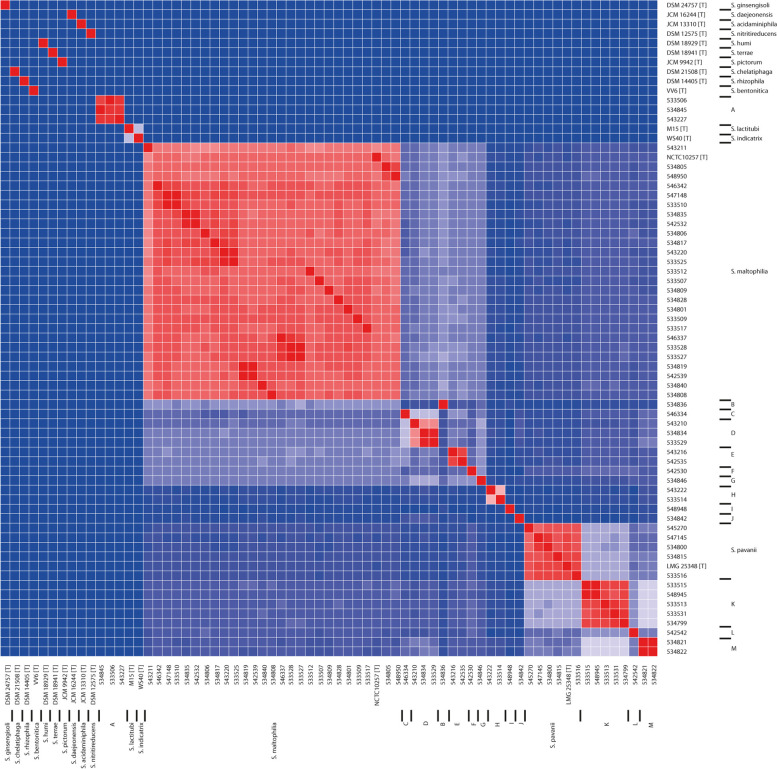
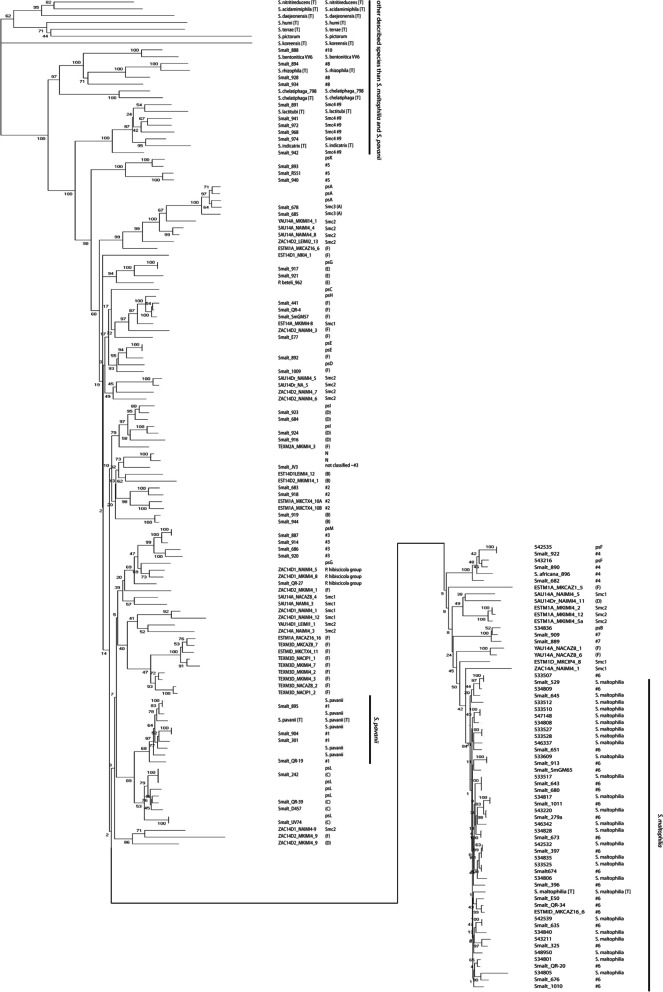
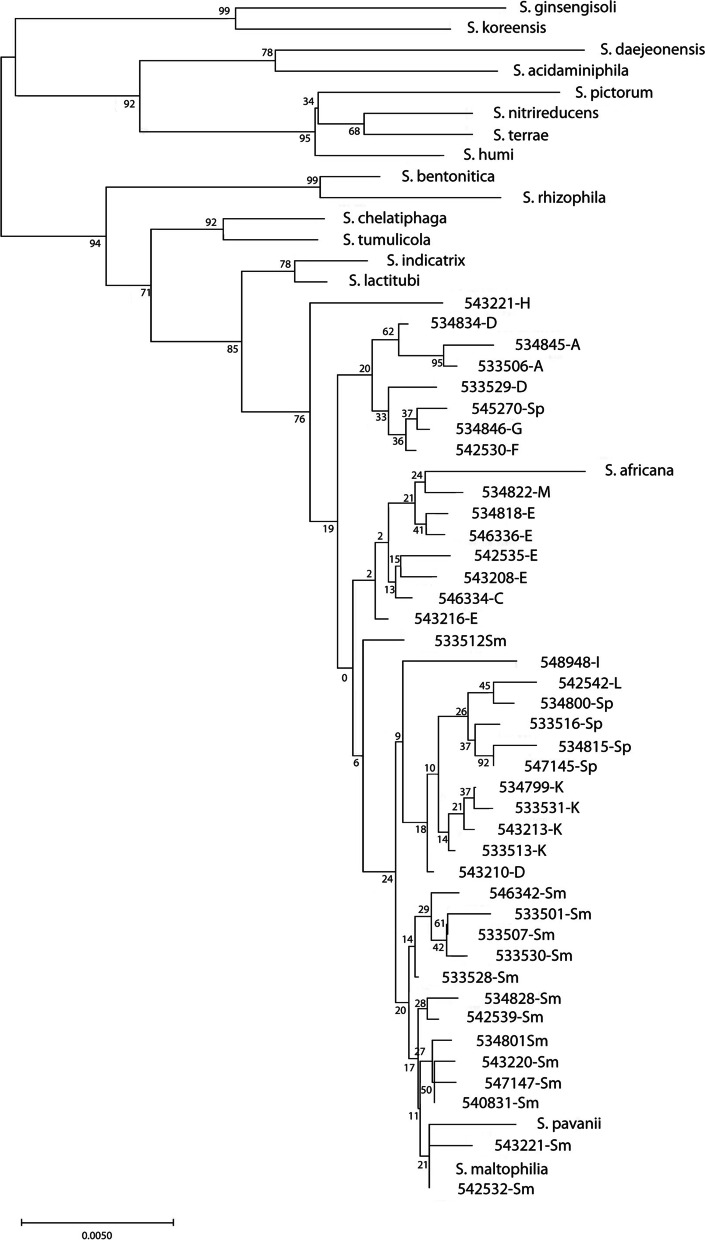
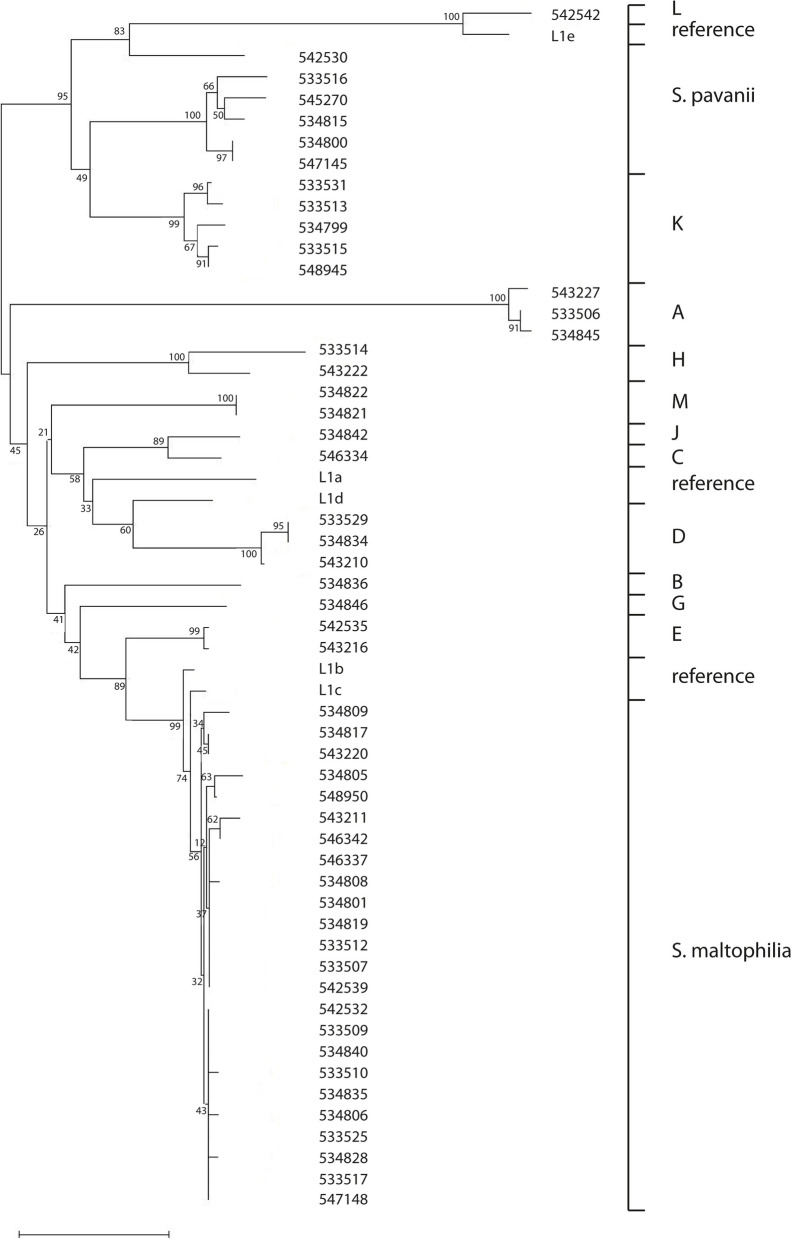
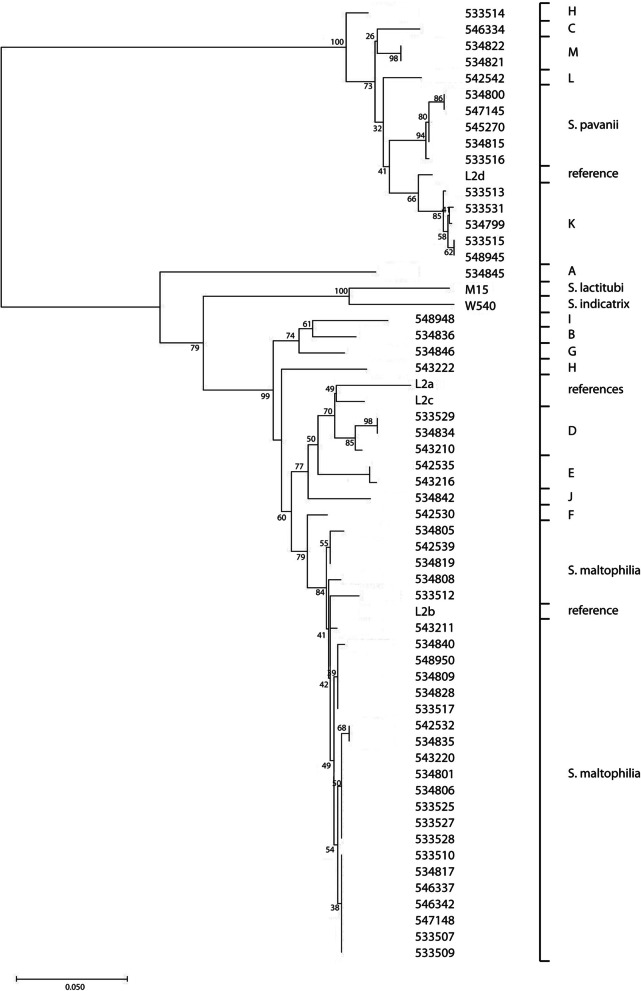
Results: A total of 111 isolates recovered between 2003 and 2016 from respiratory samples from patients in five different countries were included. Based on a cut-off of 95%, analysis of average nucleotide identity by BLAST (ANIb) showed that the 111 isolates identified as S. maltophilia by Matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF/MS) belonged to S. maltophilia (n = 65), S. pavanii (n = 6) and 13 putative novel species (n = 40), which each included 1-5 isolates; these groupings coincided with the results of the 16S rDNA analysis, and the L1 and L2 ß-lactamase Neighbor-Joining phylogeny. Chromosomally encoded aminoglycoside resistance was identified in all S. maltophilia and S. pavani isolates, while acquired antibiotic resistance genes were present in only a few isolates. Nevertheless, phenotypic resistance levels against commonly used antibiotics, determined by standard broth microbroth dilution, were high. Although putative virulence genes were present in all isolates, the percentage of positive isolates varied. The Xps II secretion system responsible for the secretion of the StmPr1-3 proteases was mainly limited to isolates identified as S. maltophilia based on ANIb, but no correlation with phenotypic expression of protease activity was found. The RPF two-component quorum sensing system involved in virulence and antibiotic resistance expression has two main variants with one variant lacking 190 amino acids in the sensing region.
Conclusions: The putative novel Stenotrophomonas species recovered from patient samples and identified by MALDI-TOF/MS as S. maltophilia, differed from S. maltophilia in resistance and virulence genes, and therefore possibly in pathogenicity. Revision of the Stenotrophomonas taxonomy is needed in order to reliably identify strains within the genus and elucidate the role of the different species in disease.
Keywords: Antibiotic resistance; Cystic fibrosis; Respiratory infection; Stenotrophomonas; Taxonomy; Virulence.
© 2022. The Author(s).
Conflict of interest statement
The authors have no competing interests to declare.
Figures
Similar articles
-
Stenotrophomonas maltophilia phenotypic and genotypic features through 4-year cystic fibrosis lung colonization.J Med Microbiol. 2021 Jan;70(1). doi: 10.1099/jmm.0.001281. Epub 2020 Nov 27. J Med Microbiol. 2021. PMID: 33258754
-
Delineation of Stenotrophomonas maltophilia isolates from cystic fibrosis patients by fatty acid methyl ester profiles and matrix-assisted laser desorption/ionization time-of-flight mass spectra using hierarchical cluster analysis and principal component analysis.J Med Microbiol. 2014 Dec;63(Pt 12):1615-1620. doi: 10.1099/jmm.0.076950-0. Epub 2014 Sep 29. J Med Microbiol. 2014. PMID: 25266870
-
Stenotrophomonas maltophilia strains from cystic fibrosis patients: genomic variability and molecular characterization of some virulence determinants.Int J Med Microbiol. 2011 Jan;301(1):34-43. doi: 10.1016/j.ijmm.2010.07.003. Epub 2010 Oct 16. Int J Med Microbiol. 2011. PMID: 20952251
-
Antibiotic treatment for Stenotrophomonas maltophilia in people with cystic fibrosis.Cochrane Database Syst Rev. 2020 Mar 18;3(3):CD009249. doi: 10.1002/14651858.CD009249.pub5. Cochrane Database Syst Rev. 2020. PMID: 32189337 Free PMC article.
-
Stenotrophomonas maltophilia: An Urgent Threat with Increasing Antibiotic Resistance.Curr Microbiol. 2023 Nov 13;81(1):6. doi: 10.1007/s00284-023-03524-5. Curr Microbiol. 2023. PMID: 37955756 Review.
Cited by
-
Phylogenomic, structural, and cell biological analyses reveal that Stenotrophomonas maltophilia replicates in acidified Rab7A-positive vacuoles of Acanthamoeba castellanii.Microbiol Spectr. 2024 Mar 5;12(3):e0298823. doi: 10.1128/spectrum.02988-23. Epub 2024 Feb 6. Microbiol Spectr. 2024. PMID: 38319117 Free PMC article.
-
Stenotrophomonas maltophilia virulence: a current view.Front Microbiol. 2024 Apr 29;15:1385631. doi: 10.3389/fmicb.2024.1385631. eCollection 2024. Front Microbiol. 2024. PMID: 38741741 Free PMC article. Review.
-
Diversity of blaL1-like genes in Stenotrophomonas species: insights from genome analysis of publicly available genome sequences.Antimicrob Agents Chemother. 2023 Sep 19;67(9):e0067323. doi: 10.1128/aac.00673-23. Epub 2023 Aug 16. Antimicrob Agents Chemother. 2023. PMID: 37584548 Free PMC article.
-
Evaluation of ethanol and EDTA concentrations in the expression of biofilm-producing smf-1, rpfF genes in XDR clinical isolates of Stenotrophomonas maltophilia.BMC Microbiol. 2023 Sep 30;23(1):277. doi: 10.1186/s12866-023-03008-3. BMC Microbiol. 2023. PMID: 37775770 Free PMC article.
-
Ceftazidime/Avibactam in Ventilator-Associated Pneumonia Due to Difficult-to-Treat Non-Fermenter Gram-Negative Bacteria in COVID-19 Patients: A Case Series and Review of the Literature.Antibiotics (Basel). 2022 Jul 26;11(8):1007. doi: 10.3390/antibiotics11081007. Antibiotics (Basel). 2022. PMID: 35892396 Free PMC article.
References
-
- List of Prokaryotic names with Standing in Nomenclature. http://www.bacterio.net/ralstonia.html. (accessed Feb 20, 2020).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
