

Using DMS-MaPseq to uncover the roles of DEAD-box proteins in ribosome assembly
- PMID: 35550176
- PMCID: PMC10152975
- DOI: 10.1016/j.ymeth.2022.05.001
Using DMS-MaPseq to uncover the roles of DEAD-box proteins in ribosome assembly
Abstract
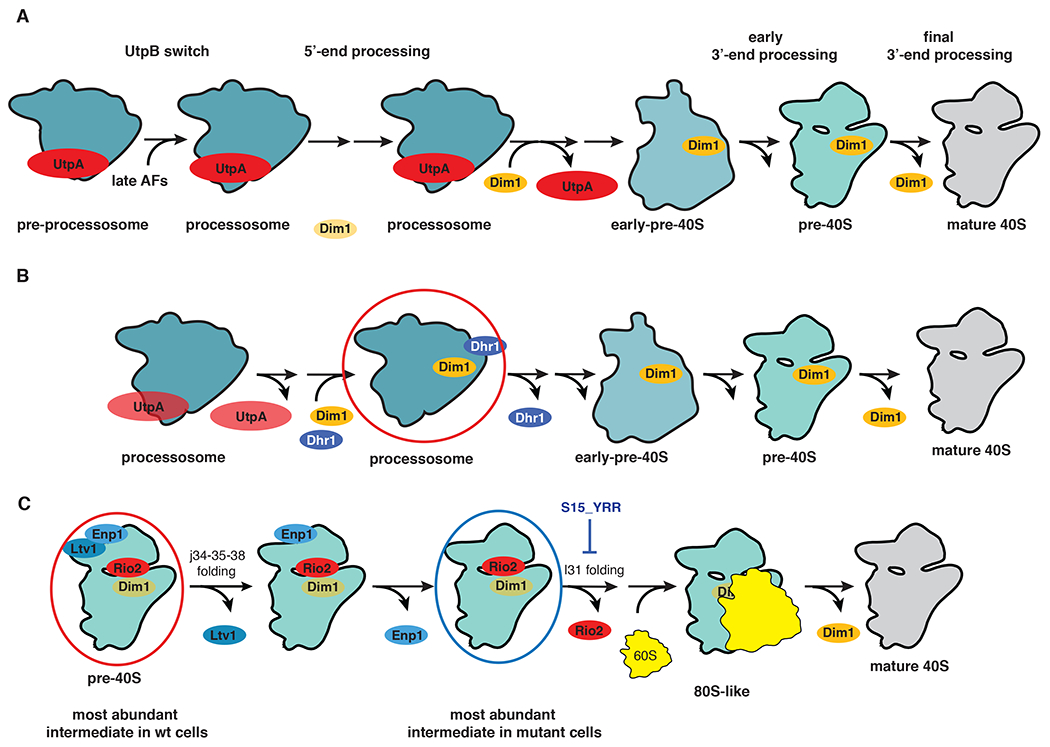
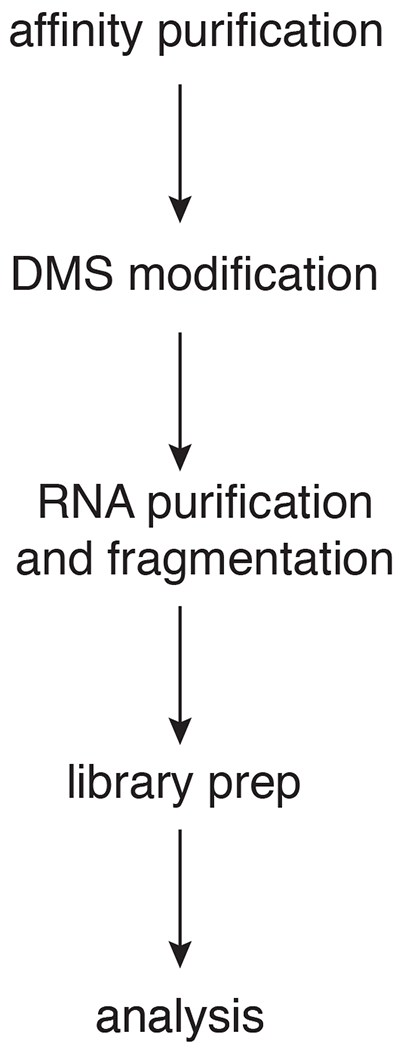
DMS (dimethylsulfate) is a time-tested chemical probe for nucleic acid secondary structure that has recently re-emerged as a powerful tool to study RNA structure and structural changes, by coupling it to high throughput sequencing techniques. This variant, termed DMS-MaPseq, allows for mapping of all RNAs in a cell at the same time. However, if an RNA adopts different structures, for example during the assembly of an RNA-protein complex, or as part of its functional cycle, then DMS-MaPseq cannot differentiate between these structures, and an ensemble average will be produced. This is especially challenging for long-lived RNAs, such as ribosomes, whose steady-state abundance far exceeds that of any assembly intermediates, rendering those inaccessible to DMS-MaPseq on total RNAs. These challenges can be overcome by purification of assembly intermediates stalled at specific assembly steps (or steps in the functional cycle), via a combination of affinity tags and mutants stalled at defined steps, and subsequent DMS probing of these intermediates. Interpretation of the differences in DMS accessibility is facilitated by additional structural information, e.g. from cryo-EM experiments, available for many functional RNAs. While this approach is generally useful for studying RNA folding or conformational changes within RNA-protein complexes, it can be particularly valuable for studying the role(s) of DEAD-box proteins, as these tend to lead to larger conformational rearrangements, often resulting from the release of an RNA-binding protein from a bound RNA. Here we provide an adaptation of the DMS-MaPseq protocol to study RNA conformational transitions during ribosome assembly, which addresses the challenges arising from the presence of many assembly intermediates, all at concentrations far below that of mature ribosomes. While this protocol was developed for the yeast S. cerevisiae, we anticipate that it should be readily transferable to other model organisms for which affinity purification has been established.
Keywords: Affinity purification; DEAD-box proteins; DMS; MapSeq; Ribosome assembly.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Moazed D, Stern S, Noller HF, Rapid chemical probing of conformation in 16 S ribosomal RNA and 30 S ribosomal subunits using primer extension, J. Mol. Biol 187(3) (1986) 399–416. - PubMed
-
- Stern S, Moazed D, Noller HF, Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension, Methods Enzymol. 164 (1988) 481–489. - PubMed
-
- Moazed D, Noller HF, Intermediate states in the movement of transfer RNA in the ribosome, Nature 342(6246) (1989) 142–8. - PubMed
-
- Mohr S, Ghanem E, Smith W, Sheeter D, Qin Y, King O, Polioudakis D, Iyer VR, Hunicke-Smith S, Swamy S, Kuersten S, Lambowitz AM, Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing, Rna 19(7) (2013) 958–70. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
