

From Wolf-Hirschhorn syndrome to NSD2 haploinsufficiency: a shifting paradigm through the description of a new case and a review of the literature
- PMID: 35550183
- PMCID: PMC9097050
- DOI: 10.1186/s13052-022-01267-w
From Wolf-Hirschhorn syndrome to NSD2 haploinsufficiency: a shifting paradigm through the description of a new case and a review of the literature
Abstract
Background: Wolf-Hirschhorn syndrome (WHS) is a well-defined disorder, whose core phenotype encompasses growth restriction, facial gestalt, intellectual disability and seizures. Nevertheless, great phenotypic variability exists due to the variable extent of the responsible 4p deletion. In addition, exome sequencing analyses, recently identified two genes, namely NSD2 and NELFA, whose loss-of-function variants contribute to a clinical spectrum consistent with atypical or partial WHS. The observation of patients exhibiting clinical features resembling WHS, with only mild developmental delay and without the typical dysmorphic features, carrying microdeletions sparing NSD2, has lead to the hypothesis that NSD2 is responsible for the intellectual disability and the facial gestalt of WHS. While presenting some of the typical findings of WHS (intellectual disability, facial gestalt, microcephaly, growth restriction and congenital heart defects), NSD2-deleted children tend to display a milder spectrum of skeletal abnormalities, usually consisting of clinodactyly, and do not exhibit seizures. We describe the clinical picture of a child with WHS due to a de novo mutation of NSD2 and discuss the clinical and diagnostic implications.
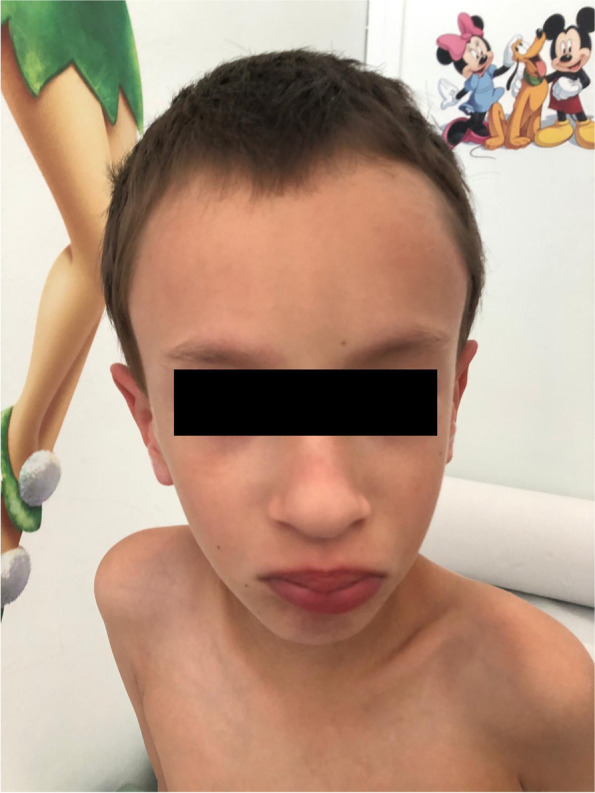
Case presentation: A 6-year-old boy was evaluated for a history of intrauterine growth restriction, low birth weight, neonatal hypotonia, and psychomotor delay. No episodes of seizure were reported. At physical examination, he displayed marphanoid habitus, muscle hypotrophy and facial dysmorphisms consisting in high frontal hairline, upslanting palpebral fissures and full lips with bifid ugula. Cryptorchidism, shawl scrotum, mild clinodactyly of the right little finger and bilateral syndactyly of the II and III toes with sandal gap were also noted. The radiographic essay demonstrated delayed bone age and echocardiography showed mild mitral prolapse. Whole genome sequencing analysis revealed a heterozygous de novo variant of NSD2 (c.2523delG).
Conclusions: Full WHS phenotype likely arises from the cumulative effect of the combined haploinsufficiency of several causative genes mapping within the 4p16.3 region, as a contiguous genes syndrome, with slightly different phenotypes depending on the specific genes involved in the deletion. When evaluating children with pictures resembling WHS, in absence of seizures, clinicians should consider this differential diagnosis.
Keywords: NSD2; Wolf-Hirschhorn syndrome; case report; facial gestalt; growth restriction; intellectual disability.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
De novo truncating variant in NSD2gene leading to atypical Wolf-Hirschhorn syndrome phenotype.BMC Med Genet. 2019 Aug 5;20(1):134. doi: 10.1186/s12881-019-0863-2. BMC Med Genet. 2019. PMID: 31382906 Free PMC article.
-
The first familial NSD2 cases with a novel variant in a Chinese father and daughter with atypical WHS facial features and a 7.5-year follow-up of growth hormone therapy.BMC Med Genomics. 2020 Dec 4;13(1):181. doi: 10.1186/s12920-020-00831-9. BMC Med Genomics. 2020. PMID: 33276791 Free PMC article.
-
Case report: A de novo NSD2 truncating variant in a child with Rauch-Steindl syndrome.Front Pediatr. 2023 Jun 7;11:1064783. doi: 10.3389/fped.2023.1064783. eCollection 2023. Front Pediatr. 2023. PMID: 37351323 Free PMC article.
-
Developmental delay and failure to thrive associated with a loss-of-function variant in WHSC1 (NSD2).Am J Med Genet A. 2018 Dec;176(12):2798-2802. doi: 10.1002/ajmg.a.40498. Epub 2018 Oct 22. Am J Med Genet A. 2018. PMID: 30345613 Review.
-
Wolf-Hirschhorn Syndrome – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY.2002 Apr 29 [updated 2015 Aug 20]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2002 Apr 29 [updated 2015 Aug 20]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301362 Free Books & Documents. Review.
Cited by
-
NSD family proteins: Rising stars as therapeutic targets.Cell Insight. 2024 Feb 3;3(2):100151. doi: 10.1016/j.cellin.2024.100151. eCollection 2024 Apr. Cell Insight. 2024. PMID: 38371593 Free PMC article. Review.
-
The role of histone methyltransferases in neurocognitive disorders associated with brain size abnormalities.Front Neurosci. 2023 Feb 10;17:989109. doi: 10.3389/fnins.2023.989109. eCollection 2023. Front Neurosci. 2023. PMID: 36845425 Free PMC article. Review.
References
-
- Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, et al. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C: Semin Med Genet. 2008;148C(4):257–269. doi: 10.1002/ajmg.c.30190. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
