

Sequence-based modeling of three-dimensional genome architecture from kilobase to chromosome scale
- PMID: 35551308
- PMCID: PMC9186125
- DOI: 10.1038/s41588-022-01065-4
Sequence-based modeling of three-dimensional genome architecture from kilobase to chromosome scale
Abstract
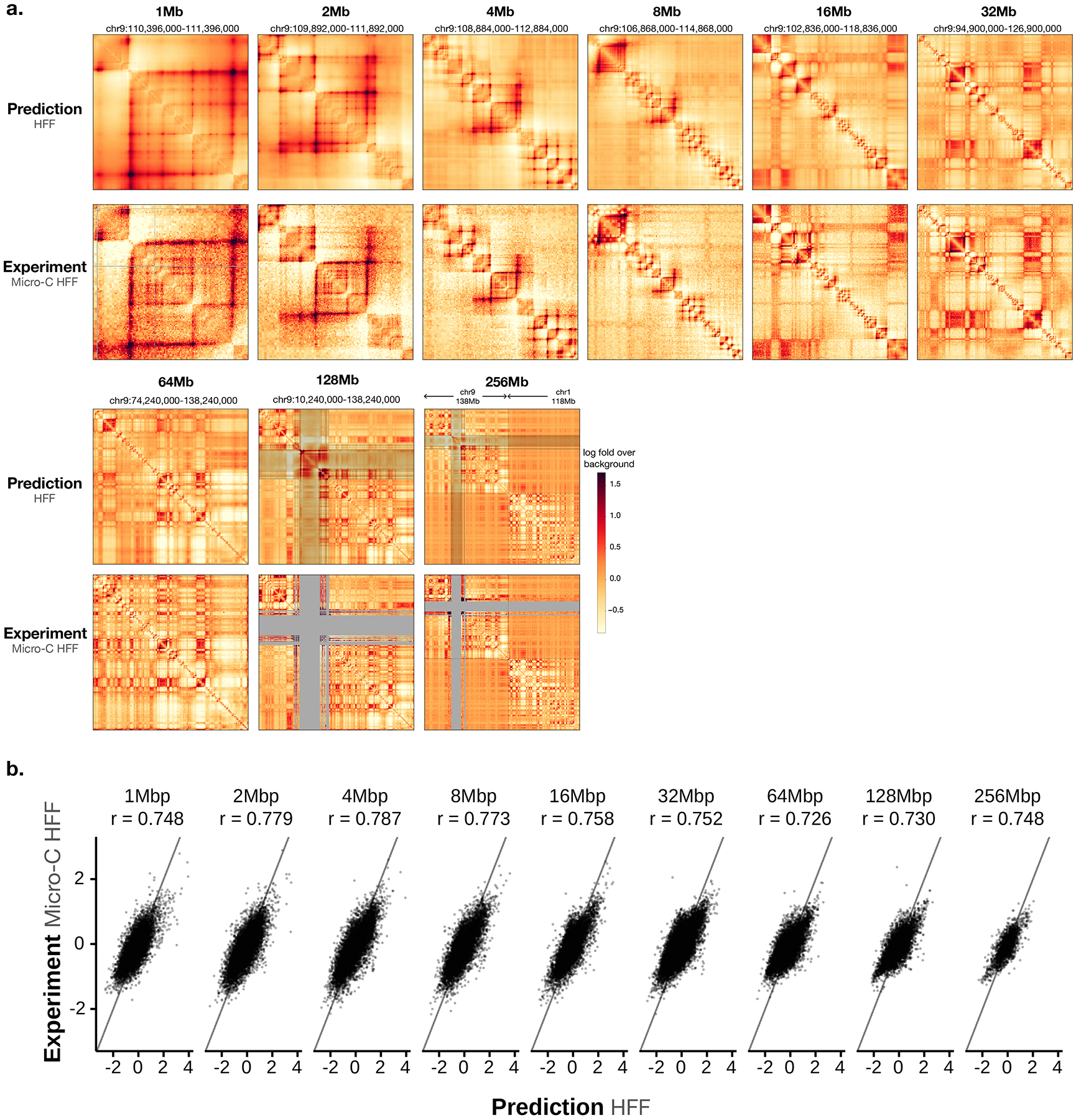
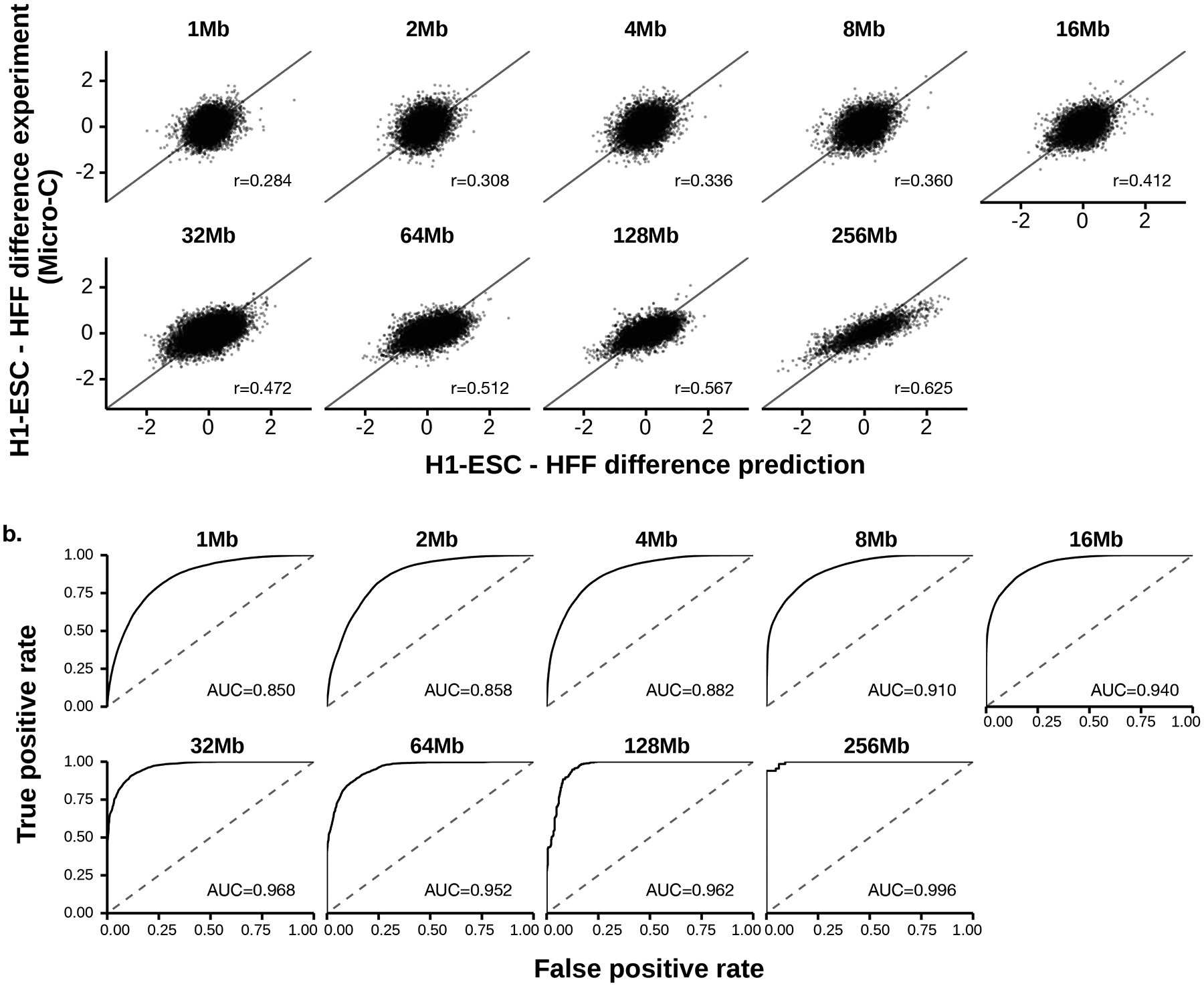
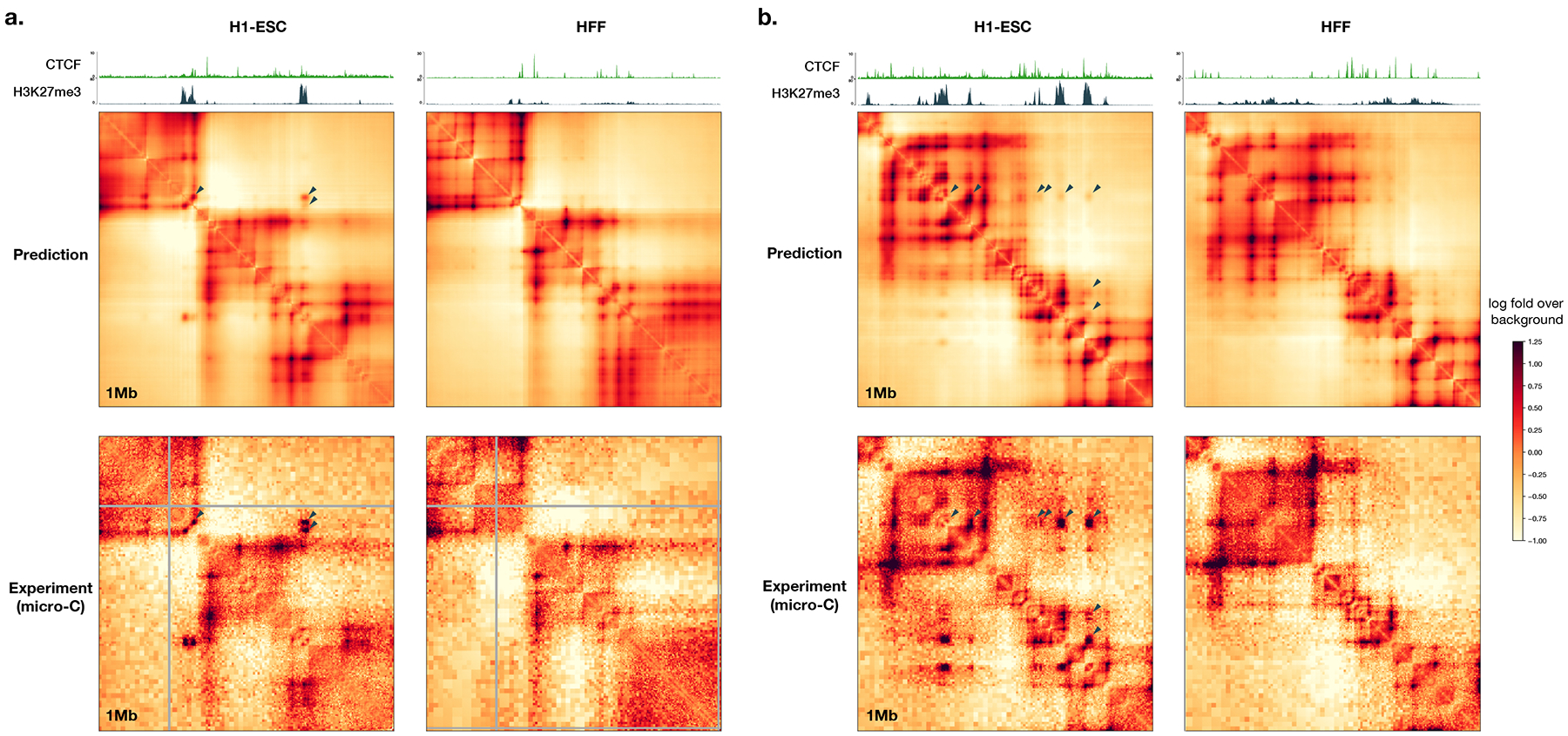
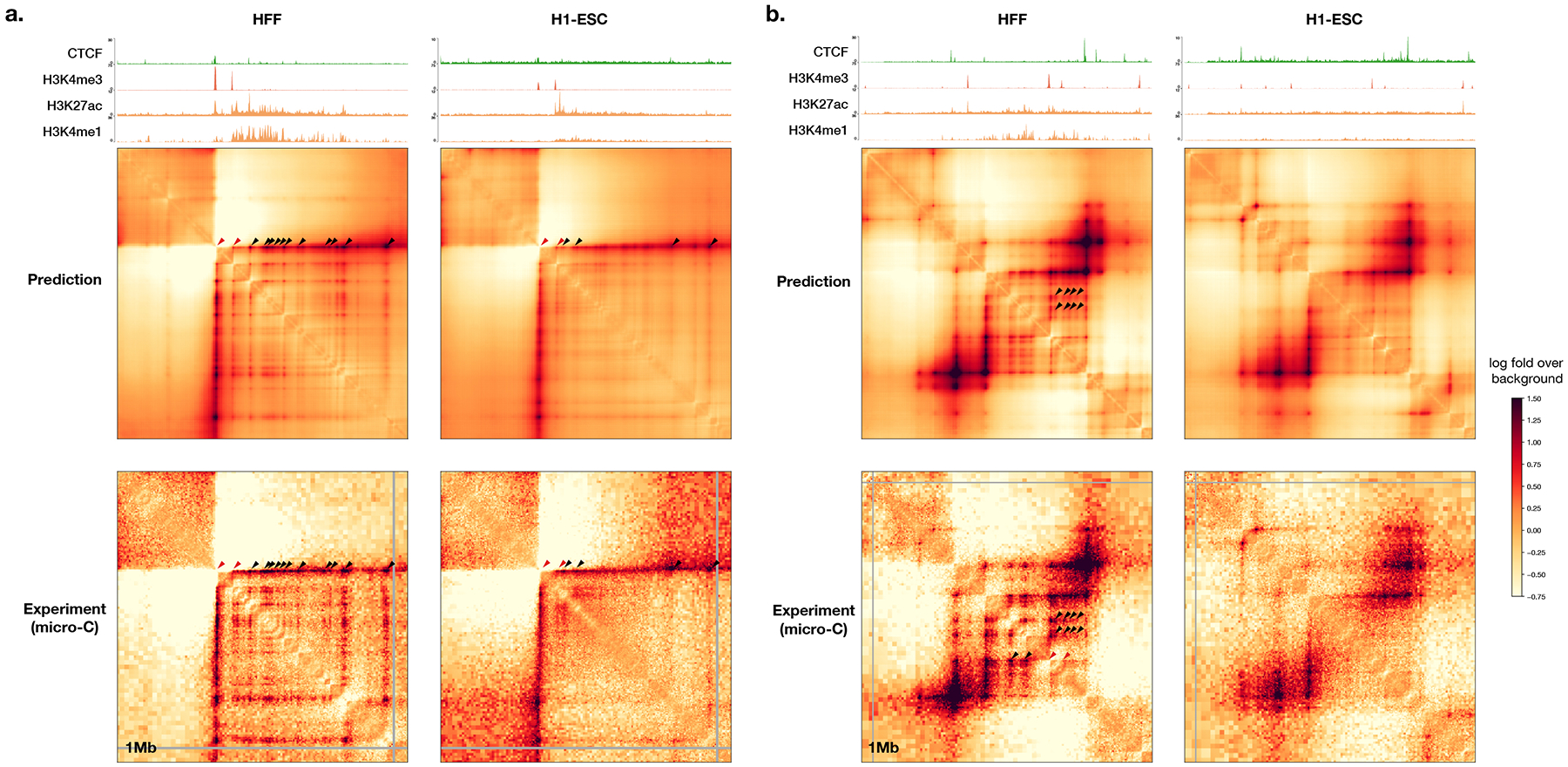
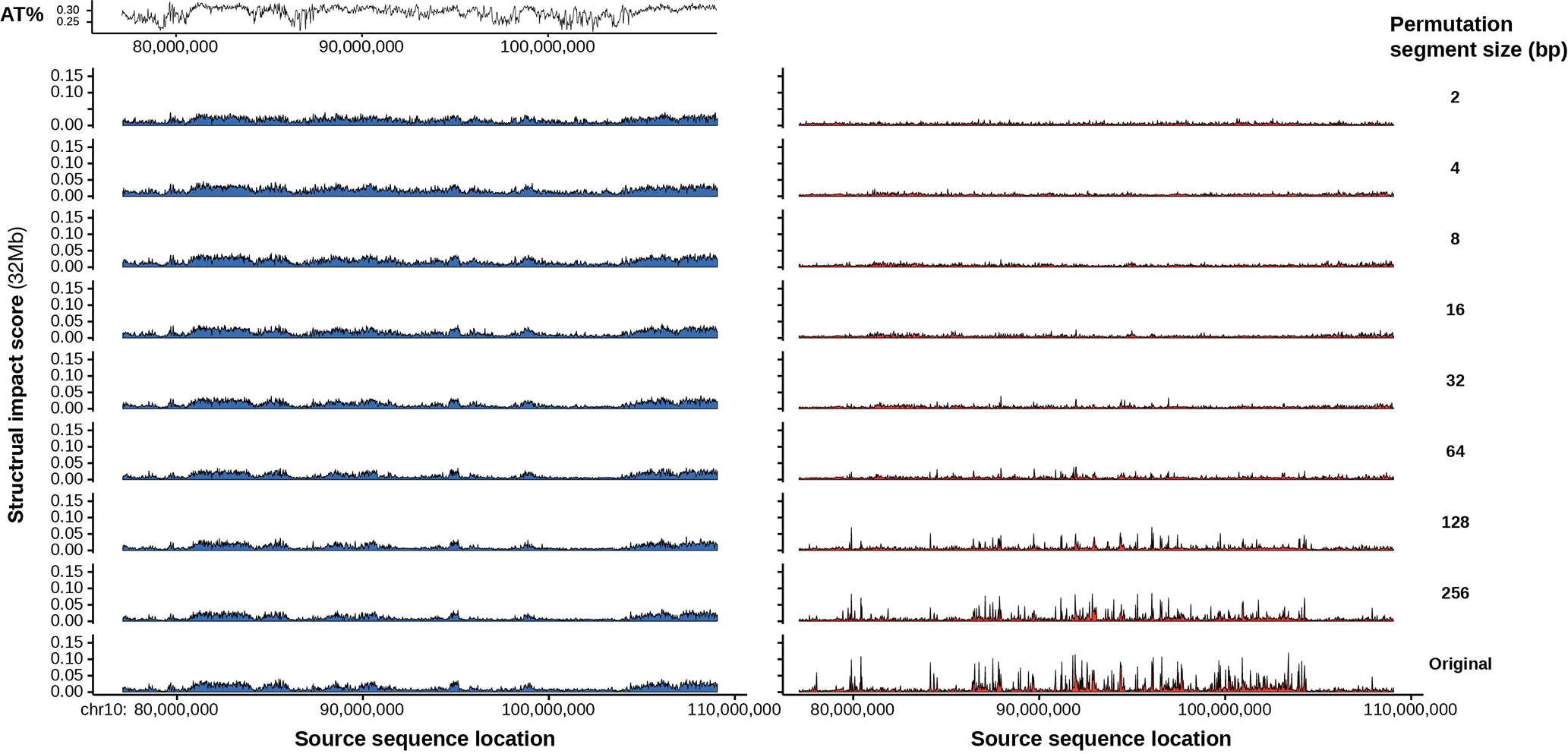
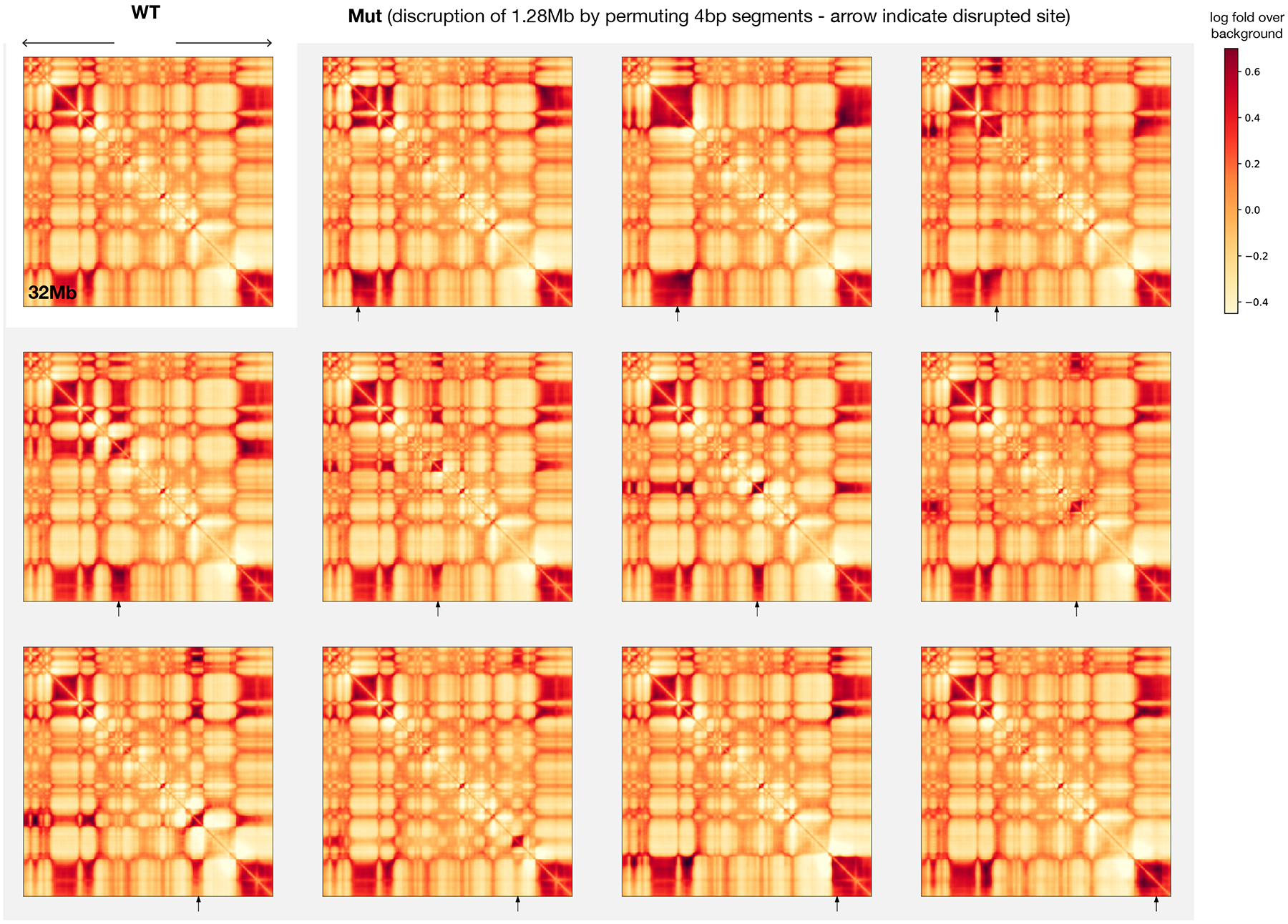
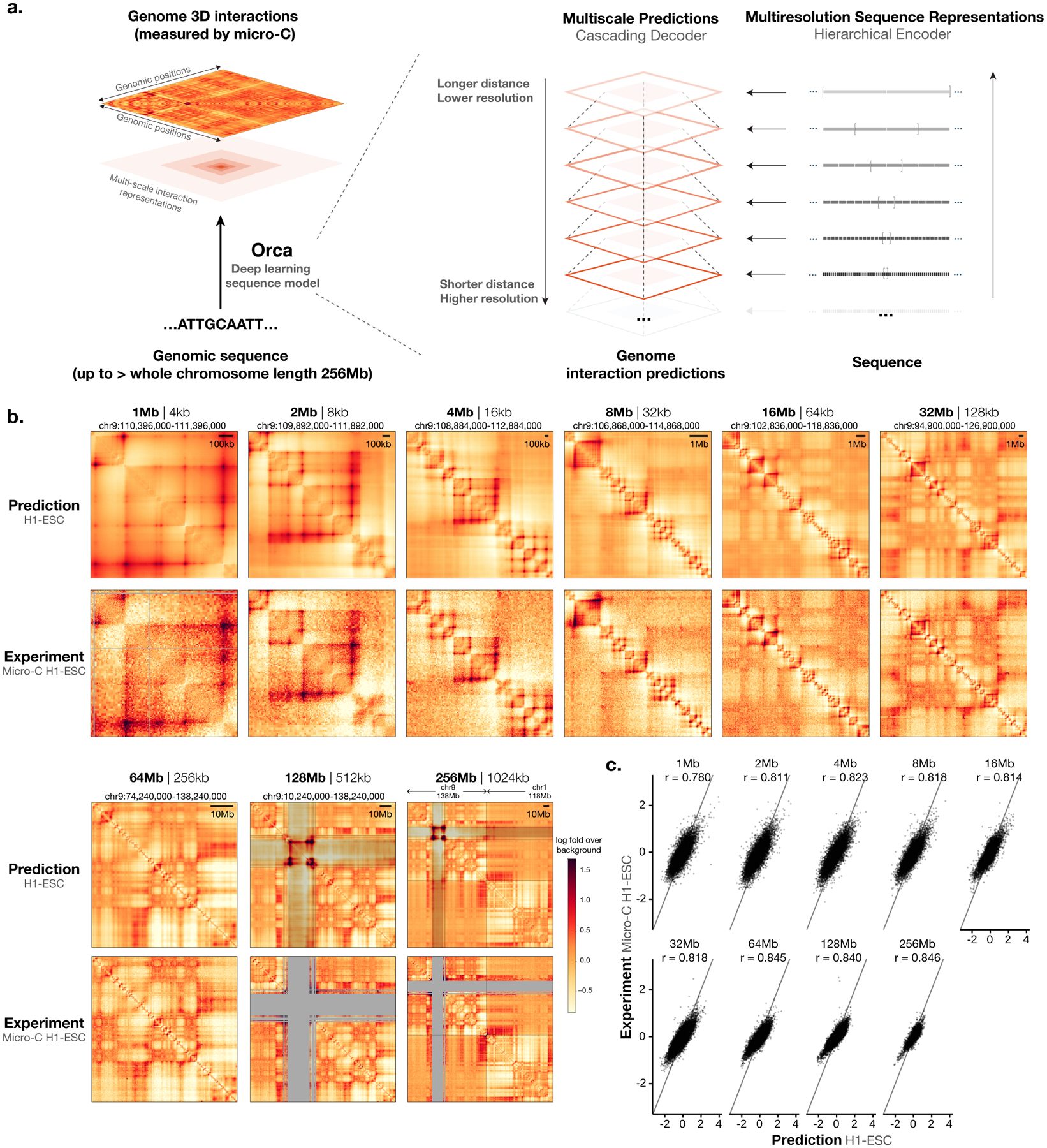
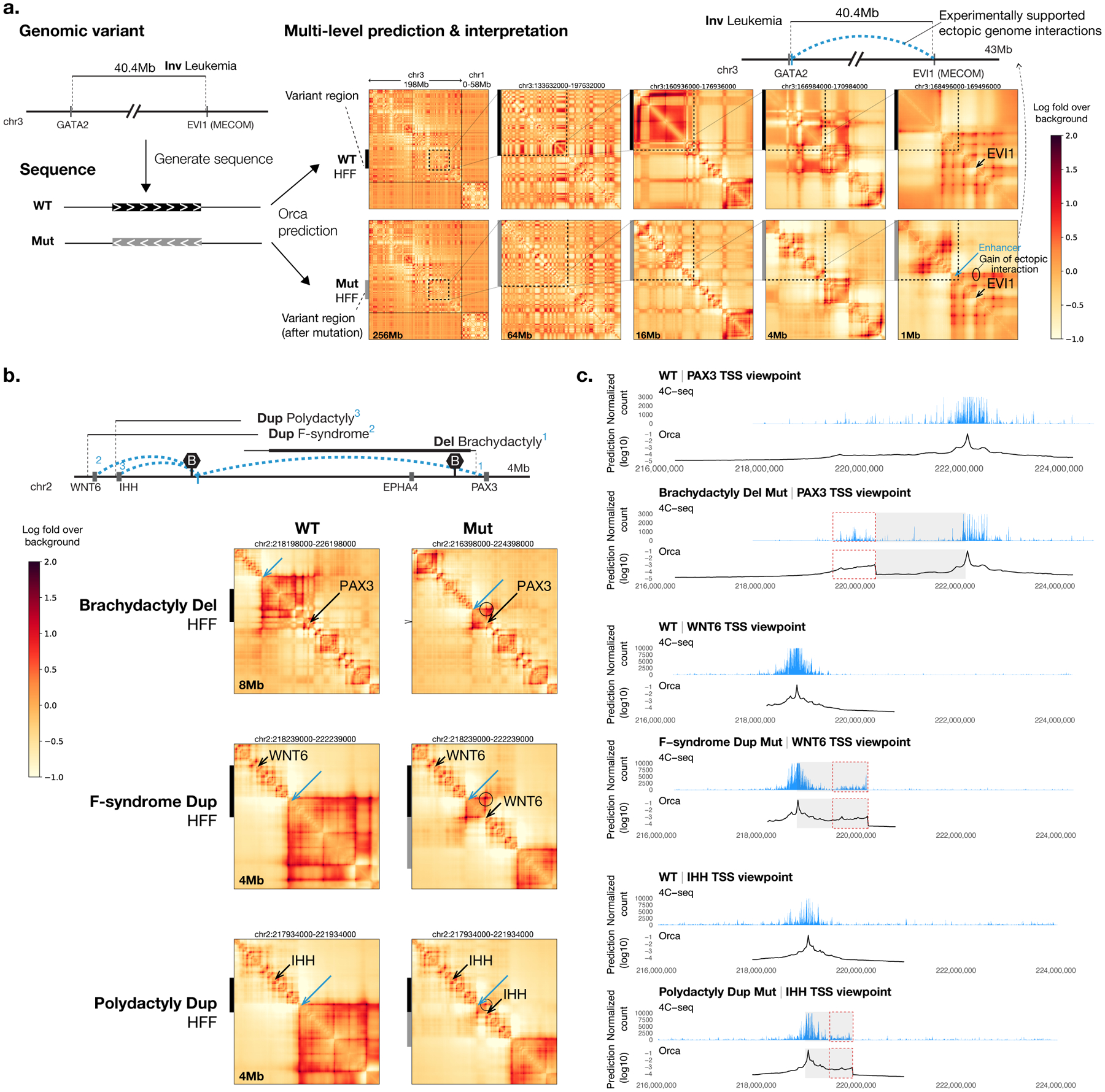
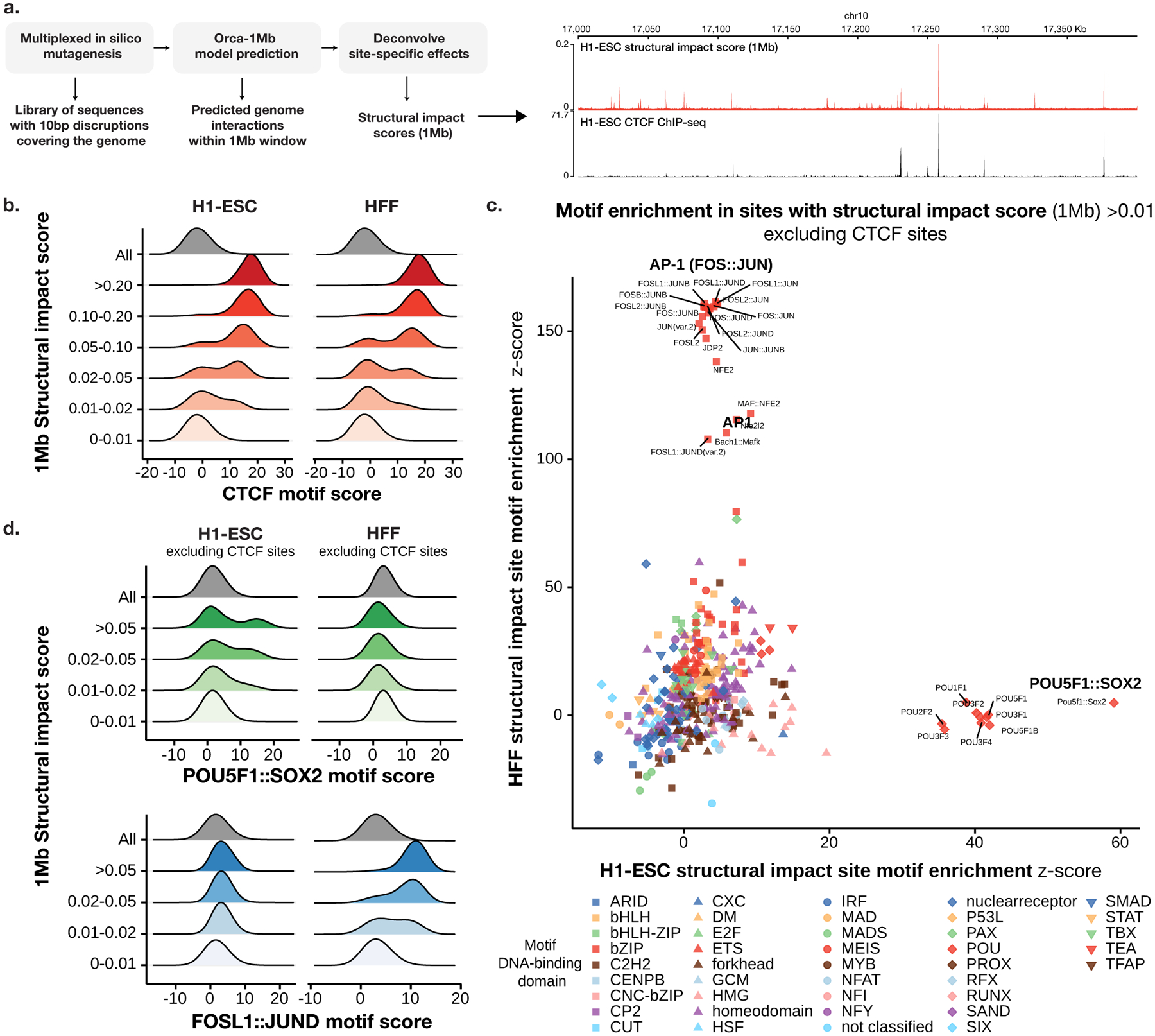
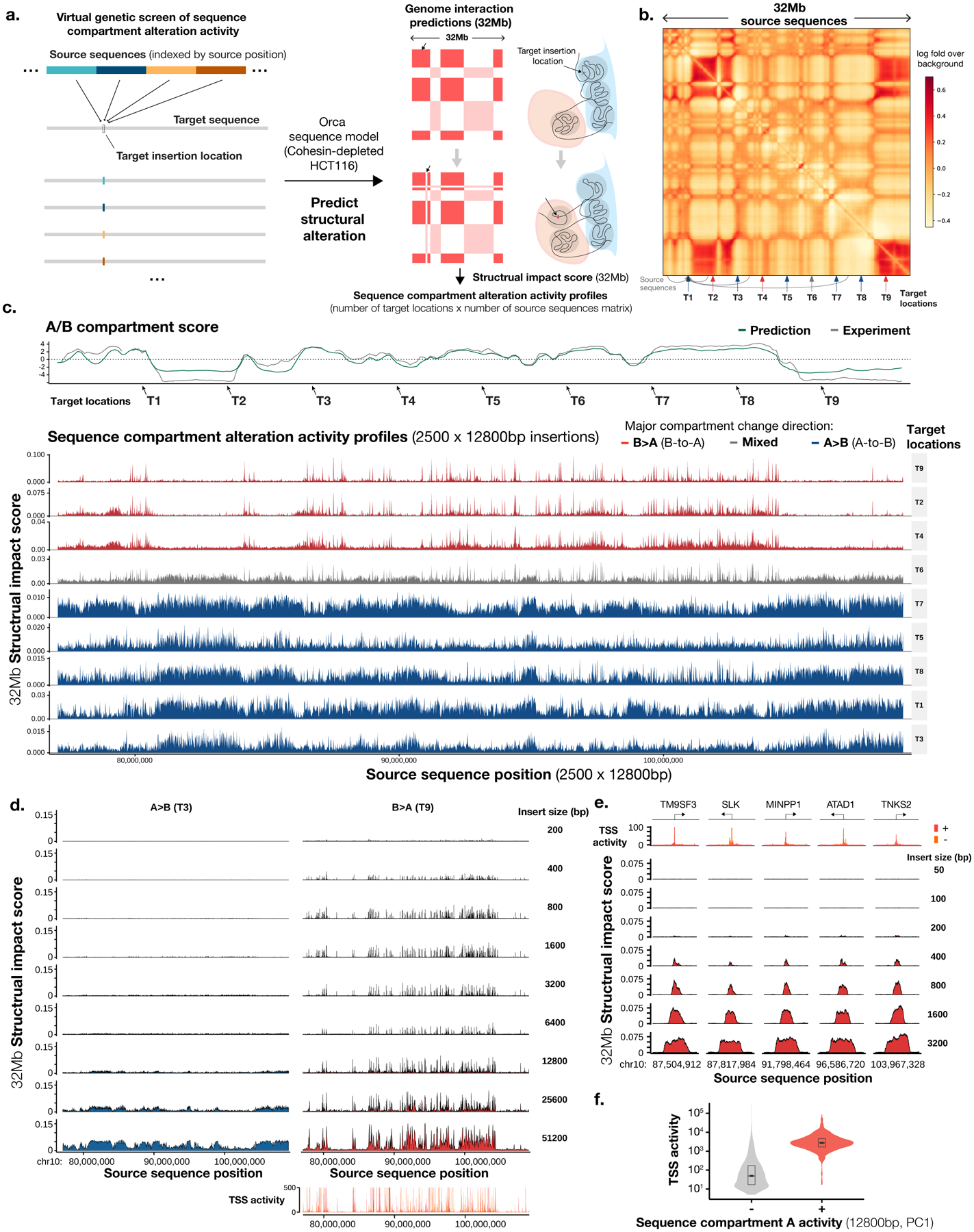
To learn how genomic sequence influences multiscale three-dimensional (3D) genome architecture, this manuscript presents a sequence-based deep-learning approach, Orca, that predicts directly from sequence the 3D genome architecture from kilobase to whole-chromosome scale. Orca captures the sequence dependencies of structures including chromatin compartments and topologically associating domains, as well as diverse types of interactions from CTCF-mediated to enhancer-promoter interactions and Polycomb-mediated interactions with cell-type specificity. Orca enables various applications including predicting structural variant effects on multiscale genome organization and it recapitulated effects of experimentally studied variants at varying sizes (300 bp to 90 Mb). Moreover, Orca enables in silico virtual screens to probe the sequence basis of 3D genome organization at different scales. At the submegabase scale, it predicted specific transcription factor motifs underlying cell-type-specific genome interactions. At the compartment scale, virtual screens of sequence activities suggest a model for the sequence basis of chromatin compartments with a prominent role of transcription start sites.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests
The author declares no competing interests.
Figures
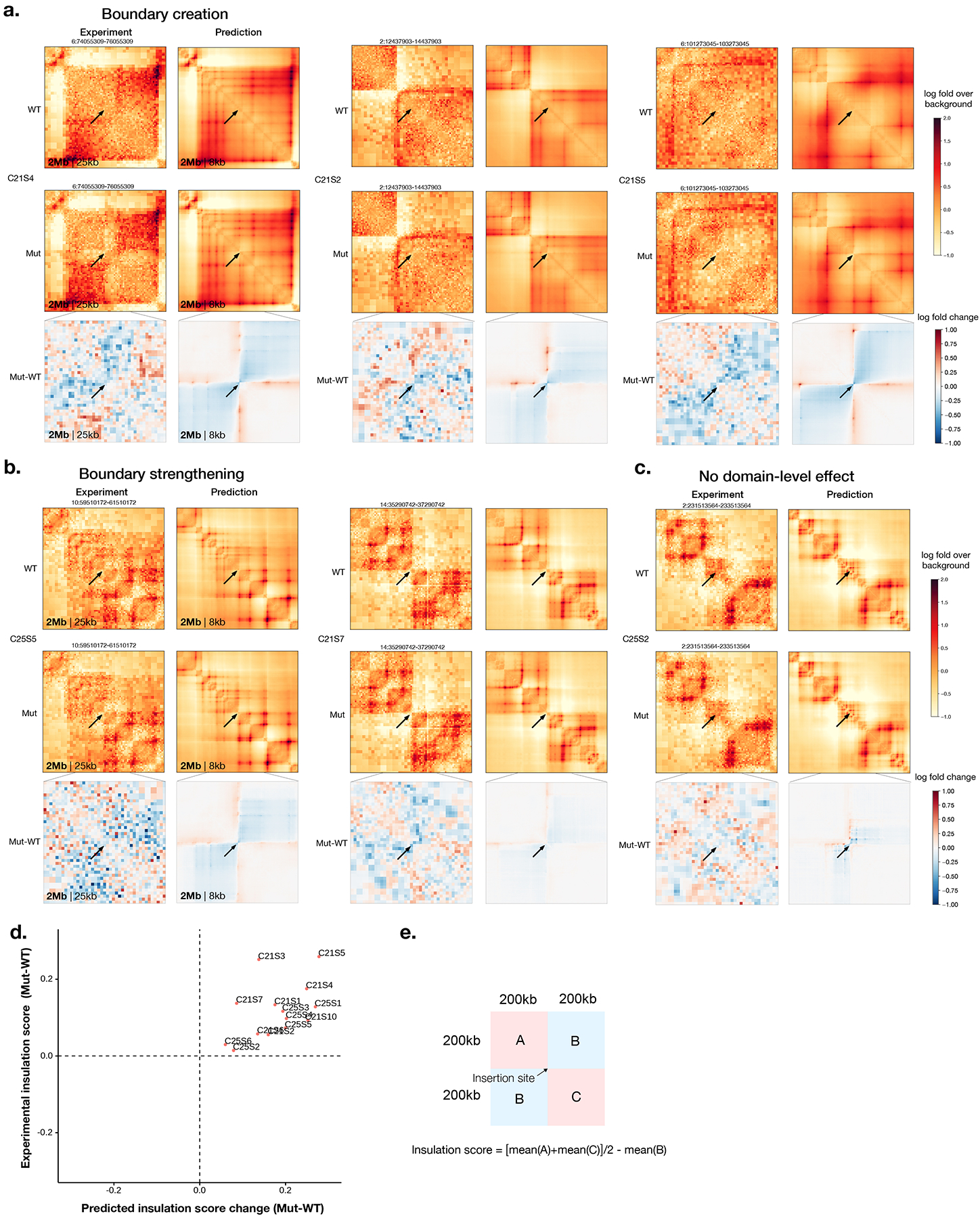
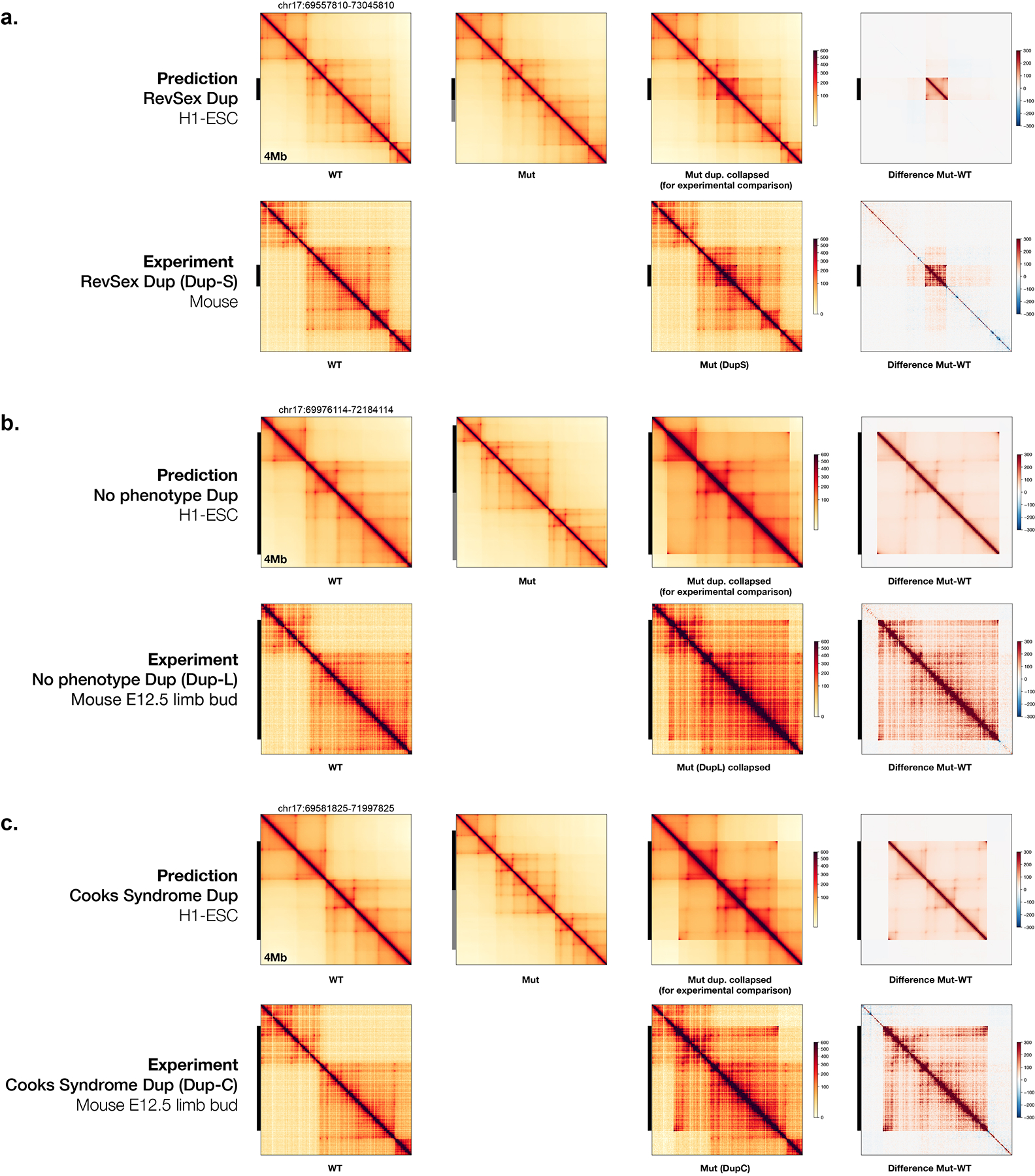
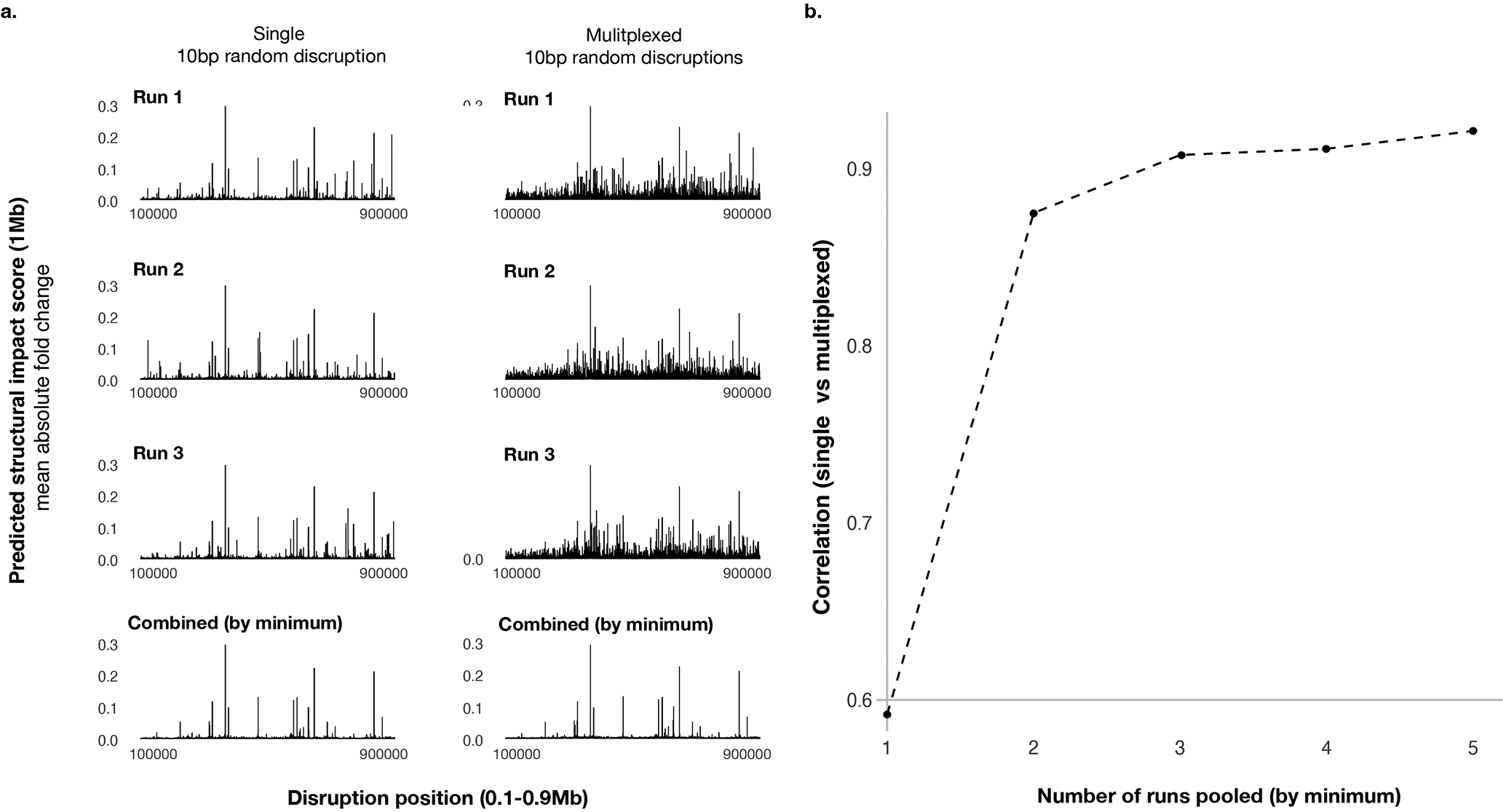
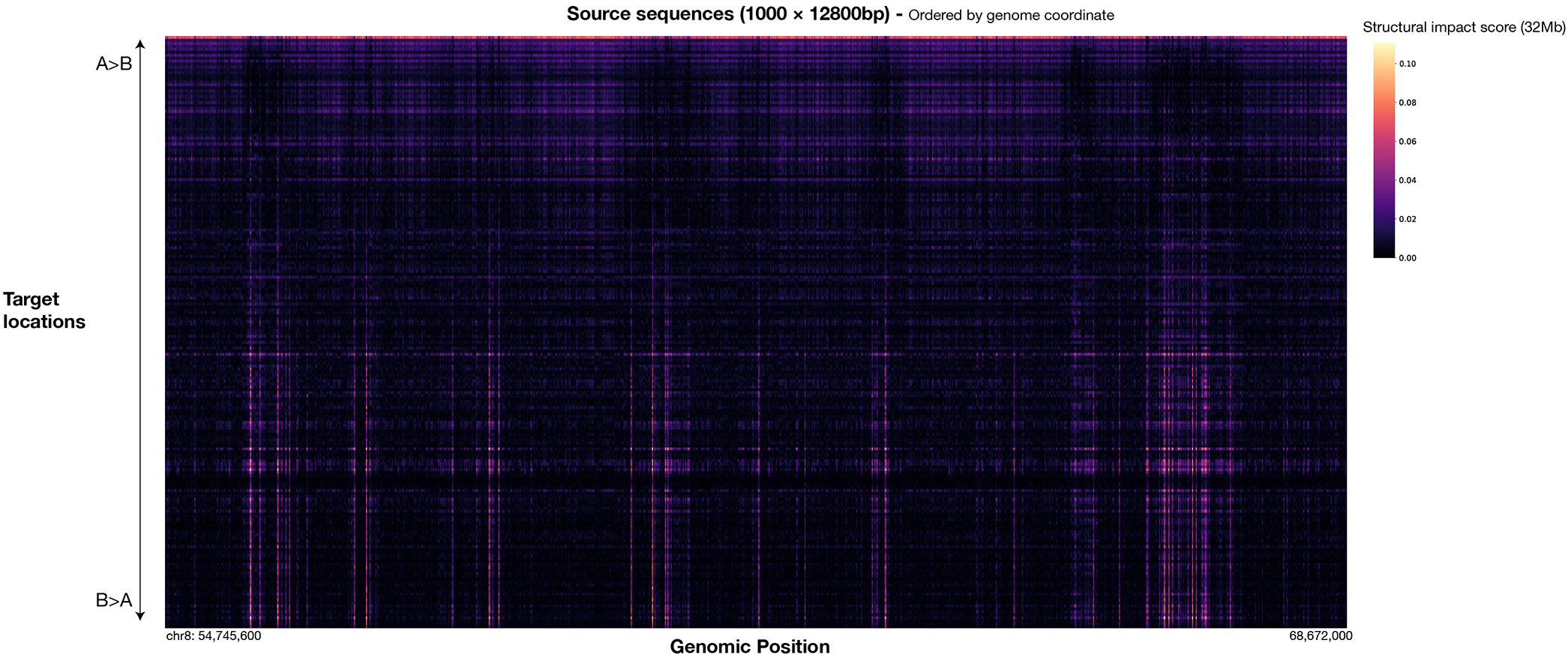
References
-
- Kosak ST et al. Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 296, 158–162 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
