

Update on Biology and Genomics of Adrenocortical Carcinomas: Rationale for Emerging Therapies
- PMID: 35551369
- PMCID: PMC9695111
- DOI: 10.1210/endrev/bnac012
Update on Biology and Genomics of Adrenocortical Carcinomas: Rationale for Emerging Therapies
Abstract
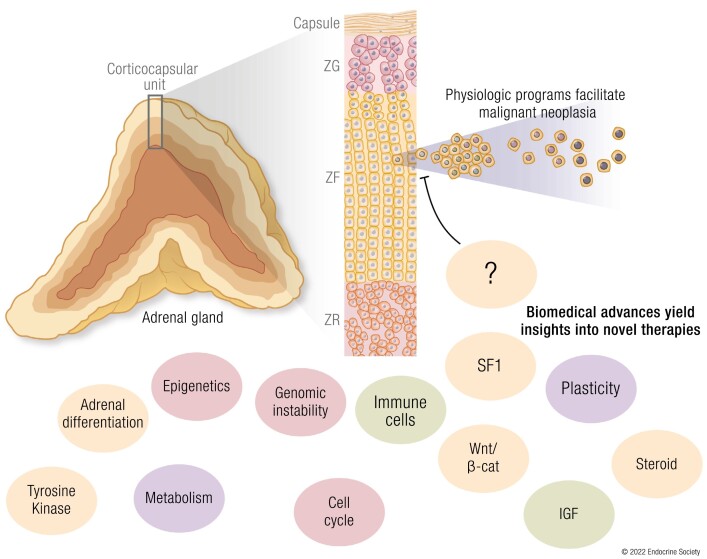
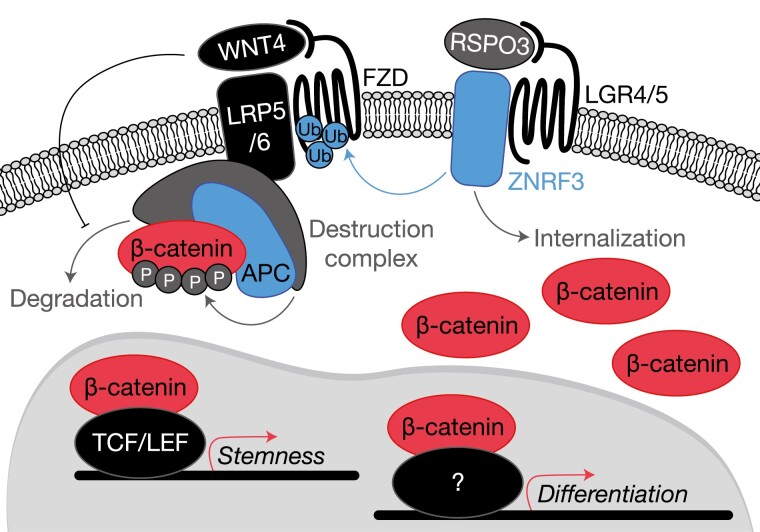
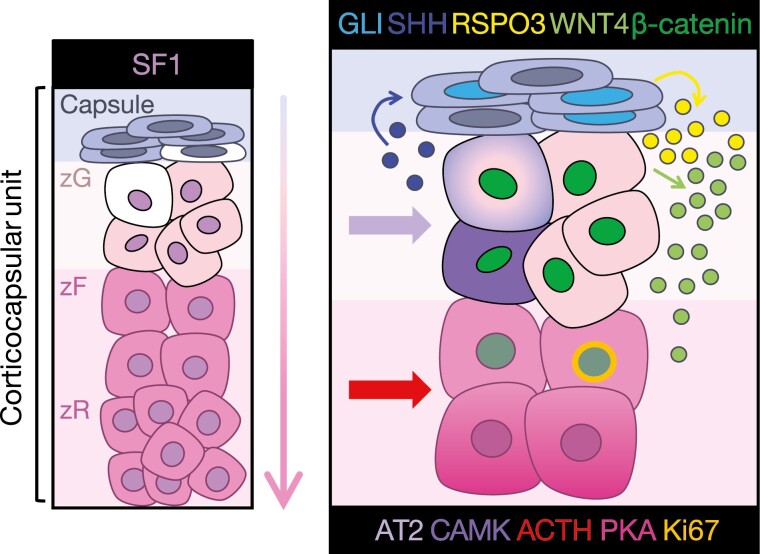
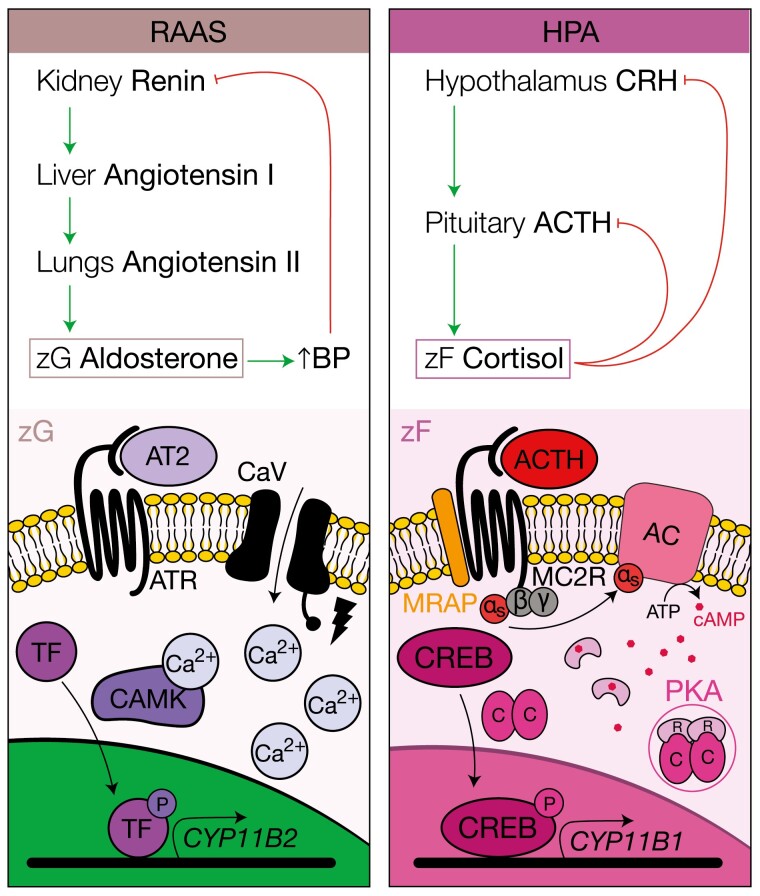
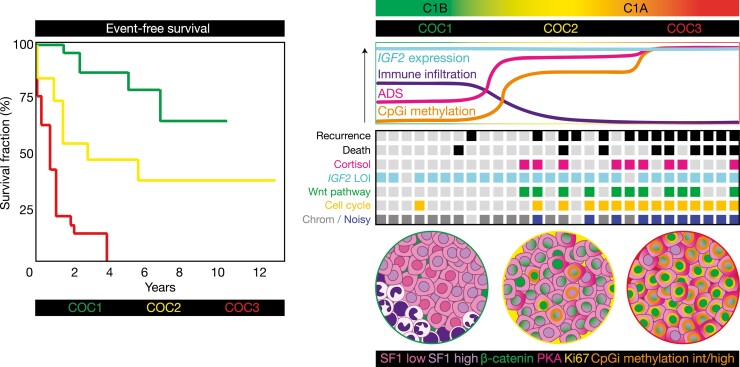
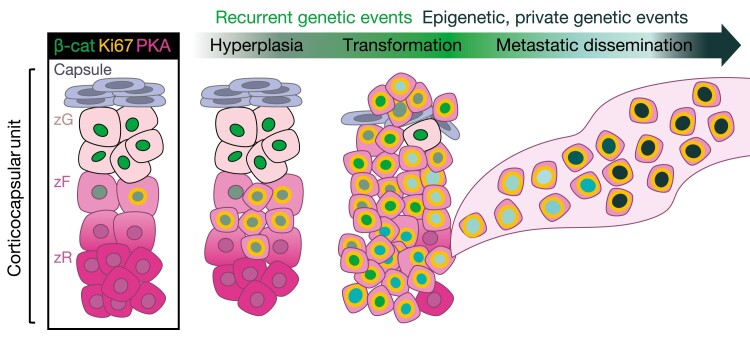
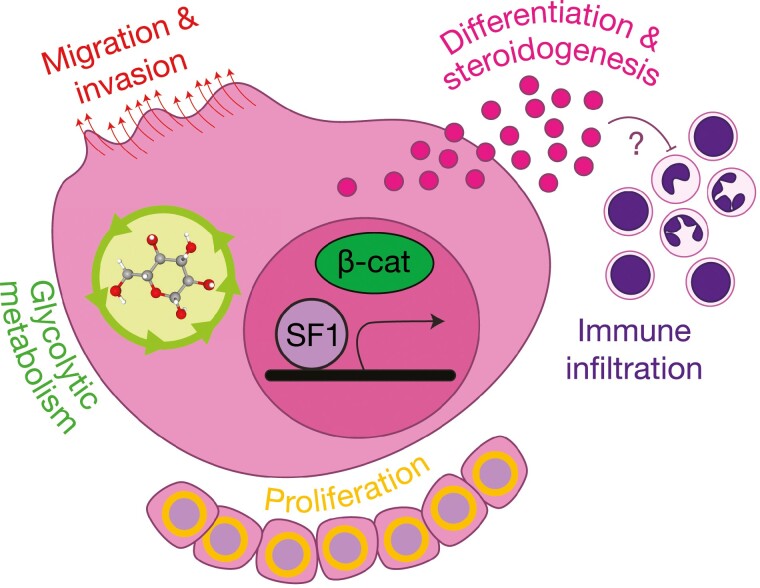
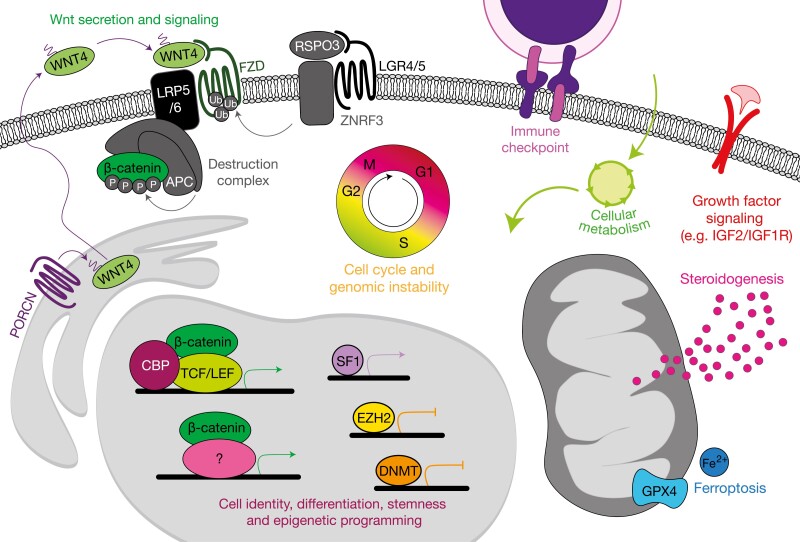
The adrenal glands are paired endocrine organs that produce steroid hormones and catecholamines required for life. Adrenocortical carcinoma (ACC) is a rare and often fatal cancer of the peripheral domain of the gland, the adrenal cortex. Recent research in adrenal development, homeostasis, and disease have refined our understanding of the cellular and molecular programs controlling cortical growth and renewal, uncovering crucial clues into how physiologic programs are hijacked in early and late stages of malignant neoplasia. Alongside these studies, genome-wide approaches to examine adrenocortical tumors have transformed our understanding of ACC biology, and revealed that ACC is composed of distinct molecular subtypes associated with favorable, intermediate, and dismal clinical outcomes. The homogeneous transcriptional and epigenetic programs prevailing in each ACC subtype suggest likely susceptibility to any of a plethora of existing and novel targeted agents, with the caveat that therapeutic response may ultimately be limited by cancer cell plasticity. Despite enormous biomedical research advances in the last decade, the only potentially curative therapy for ACC to date is primary surgical resection, and up to 75% of patients will develop metastatic disease refractory to standard-of-care adjuvant mitotane and cytotoxic chemotherapy. A comprehensive, integrated, and current bench-to-bedside understanding of our field's investigations into adrenocortical physiology and neoplasia is crucial to developing novel clinical tools and approaches to equip the one-in-a-million patient fighting this devastating disease.
Keywords: adrenocortical carcinoma; adrenocortical development and homeostasis; genomics; molecular biomarkers; targeted therapies.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
Similar articles
-
Update in adrenocortical carcinoma.J Clin Endocrinol Metab. 2013 Dec;98(12):4551-64. doi: 10.1210/jc.2013-3020. Epub 2013 Sep 30. J Clin Endocrinol Metab. 2013. PMID: 24081734 Review.
-
Mitotane Inhibits Sterol-O-Acyl Transferase 1 Triggering Lipid-Mediated Endoplasmic Reticulum Stress and Apoptosis in Adrenocortical Carcinoma Cells.Endocrinology. 2015 Nov;156(11):3895-908. doi: 10.1210/en.2015-1367. Epub 2015 Aug 25. Endocrinology. 2015. PMID: 26305886
-
Surgical management of adrenocortical carcinoma.Endocrinol Metab Clin North Am. 2015 Jun;44(2):435-52. doi: 10.1016/j.ecl.2015.02.008. Epub 2015 Mar 17. Endocrinol Metab Clin North Am. 2015. PMID: 26038210 Review.
-
European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors.Eur J Endocrinol. 2018 Oct 1;179(4):G1-G46. doi: 10.1530/EJE-18-0608. Eur J Endocrinol. 2018. PMID: 30299884
-
microRNA-431 as a Chemosensitizer and Potentiator of Drug Activity in Adrenocortical Carcinoma.Oncologist. 2019 Jun;24(6):e241-e250. doi: 10.1634/theoncologist.2018-0849. Epub 2019 Mar 27. Oncologist. 2019. PMID: 30918109 Free PMC article.
Cited by
-
Therapeutic strategies for adrenocortical carcinoma: integrating genomic insights, molecular targeting, and immunotherapy.Front Immunol. 2025 Mar 12;16:1545012. doi: 10.3389/fimmu.2025.1545012. eCollection 2025. Front Immunol. 2025. PMID: 40145087 Free PMC article. Review.
-
Molecular Subtypes Defined by Cuproptosis-Associated Genes, Prognostic Model Development, and Tumor Immune Microenvironment Characterization in Adrenocortical Carcinoma.J Inflamm Res. 2024 Oct 3;17:7017-7036. doi: 10.2147/JIR.S461489. eCollection 2024. J Inflamm Res. 2024. PMID: 39377045 Free PMC article.
-
Bilateral Adrenal Tumors: A Visual Case Series.AACE Clin Case Rep. 2024 Nov 27;11(1):79-86. doi: 10.1016/j.aace.2024.11.006. eCollection 2025 Jan-Feb. AACE Clin Case Rep. 2024. PMID: 39896942 Free PMC article. No abstract available.
-
A Case of Severe Cushing Syndrome due to Metastatic Adrenocortical Carcinoma Treated With Osilodrostat.AACE Clin Case Rep. 2024 Oct 23;11(1):53-57. doi: 10.1016/j.aace.2024.10.005. eCollection 2025 Jan-Feb. AACE Clin Case Rep. 2024. PMID: 39896957 Free PMC article.
-
The transcription factor HHEX maintains glucocorticoid levels and protects adrenals from androgen-induced lipid depletion.Res Sq [Preprint]. 2025 Apr 15:rs.3.rs-6248794. doi: 10.21203/rs.3.rs-6248794/v1. Res Sq. 2025. PMID: 40321776 Free PMC article. Preprint.
References
-
- Terzolo M, Angeli A, Fassnacht M, et al. . Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007;356(23):2372-2380. - PubMed
-
- Fassnacht M, Dekkers OM, Else T, et al. . European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2018;179(4):G1-G46. - PubMed
-
- Glenn JA, Else T, Hughes DT, et al. . Longitudinal patterns of recurrence in patients with adrenocortical carcinoma. Surgery. 2019;165(1):186-195. - PubMed
