

Analysis of clinical characteristics of children with Aicardi-Goutieres syndrome in China
- PMID: 35551623
- PMCID: PMC9205831
- DOI: 10.1007/s12519-022-00545-1
Analysis of clinical characteristics of children with Aicardi-Goutieres syndrome in China
Abstract
Background: Aicardi-Goutieres syndrome (AGS) is an inflammatory disorder belonging to the type I interferonopathy group. The clinical diagnosis of AGS is difficult, which can lead to a high mortality rate. Overall, there is a lack of large-sample research data on AGS in China. We aim to summarize the clinical characteristics of Chinese patients with AGS and provide clues for clinical diagnostic.
Methods: The genetic and clinical features of Chinese patients with AGS were collected. Real-time polymerase chain reaction was used to detect expression of interferon-stimulated genes (ISGs).
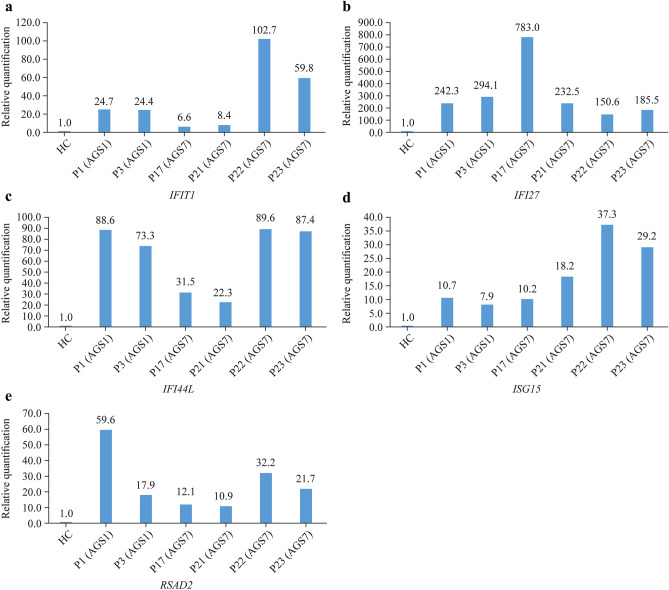
Results: A total of 23 cases were included, consisting of 7 cases of AGS1 with three prime repair exonuclease 1 mutations, 3 of AGS2 with ribonuclease H2 subunit B (RNASEH2B) mutations, 3 of ASG3 with RNASEH2C, 1 of AGS4 with RNASEH2A mutations, 2 of AGS6 with adenosine deaminase acting on RNA 1 mutations, and 7 of AGS7 with interferon induced with helicase C domain 1 mutations. Onset before the age of 3 years occurred in 82.6%. Neurologic involvement was most common (100%), including signs of intracranial calcification which mainly distributed in the bilateral basal ganglia, leukodystrophy, dystonia, epilepsy, brain atrophy and dysphagia. Intellectual disability, language disability and motor skill impairment were also observed. Skin manifestations (60.87%) were dominated by a chilblain-like rash. Features such as microcephaly (47.62%), short stature (52.38%), liver dysfunction (42.11%), thyroid dysfunction (46.15%), positive autoimmune antibodies (66.67%), and elevated erythrocyte sedimentation rate (53.85%) were also found. The phenotypes of 2 cases fulfilled the diagnostic criteria for systemic lupus erythaematosus (SLE). One death was recorded. ISGs expression were elevated.
Conclusions: AGS is a systemic disease that causes sequelae and mortality. A diagnosis of AGS should be considered for patients who have an early onset of chilblain-like rash, intracranial calcification, leukodystrophy, dystonia, developmental delay, positive autoimmune antibodies, and elevated ISGs, and for those diagnosed with SLE with atypical presentation who are nonresponsive to conventional treatments. Comprehensive assessment of vital organ function and symptomatic treatment are important.
Keywords: Aicardi-Goutieres syndrome; Chinese; Diagnosis; Manifestations.
© 2022. The Author(s).
Conflict of interest statement
Author SHM is a member of the Editorial Board for World Journal of Pediatrics. The paper was handled by the other Editor and has undergone rigorous peer review process. Author SHM was not involved in the journal's review of, or decisions related to, this manuscript. No financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article. The authors have no conflicts of interest to disclose.
Figures
References
-
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;167A:296–312. doi: 10.1002/ajmg.a.36887. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
