

Clinical and genetic spectrums of 413 North African families with inherited retinal dystrophies and optic neuropathies
- PMID: 35551639
- PMCID: PMC9097391
- DOI: 10.1186/s13023-022-02340-7
Clinical and genetic spectrums of 413 North African families with inherited retinal dystrophies and optic neuropathies
Abstract
Background: Inherited retinal dystrophies (IRD) and optic neuropathies (ION) are the two major causes world-wide of early visual impairment, frequently leading to legal blindness. These two groups of pathologies are highly heterogeneous and require combined clinical and molecular diagnoses to be securely identified. Exact epidemiological studies are lacking in North Africa, and genetic studies of IRD and ION individuals are often limited to case reports or to some families that migrated to the rest of the world. In order to improve the knowledge of their clinical and genetic spectrums in North Africa, we reviewed published data, to illustrate the most prevalent pathologies, genes and mutations encountered in this geographical region, extending from Morocco to Egypt, comprising 200 million inhabitants.
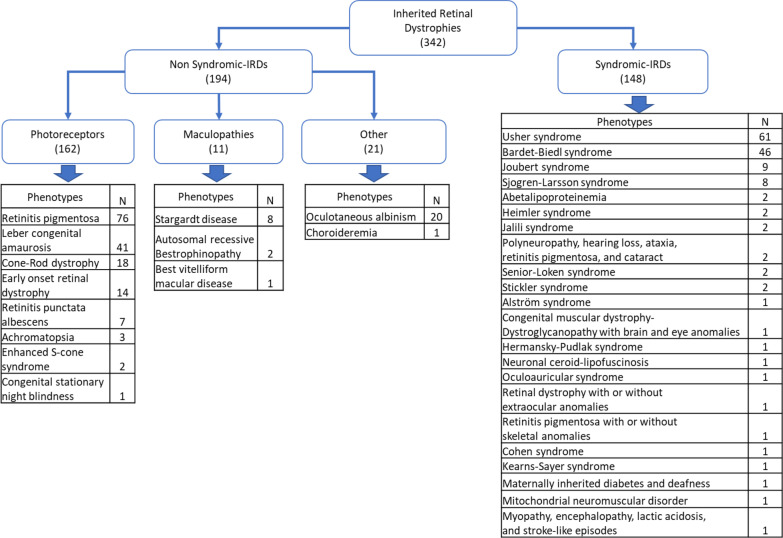
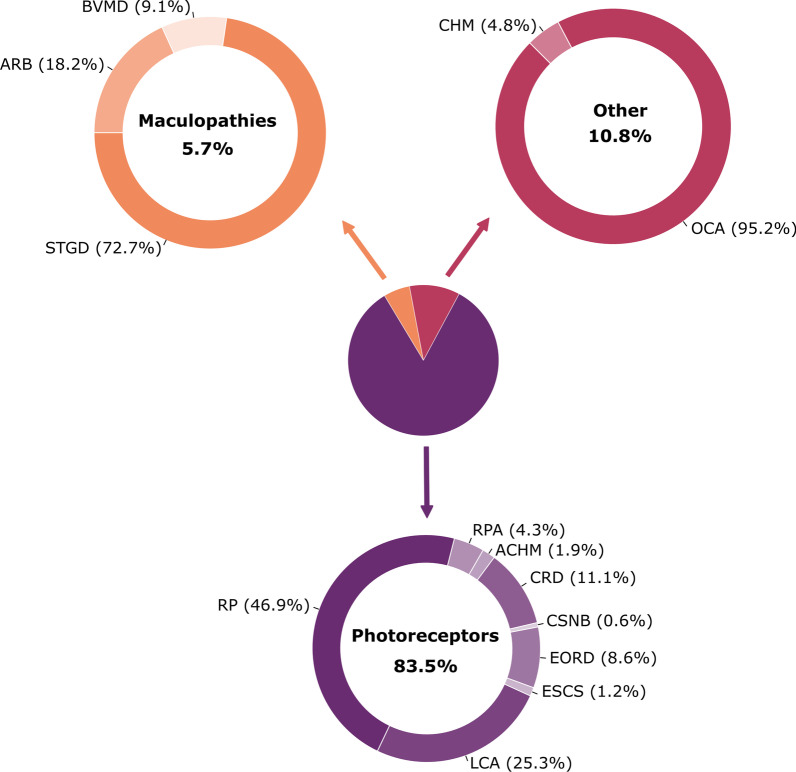
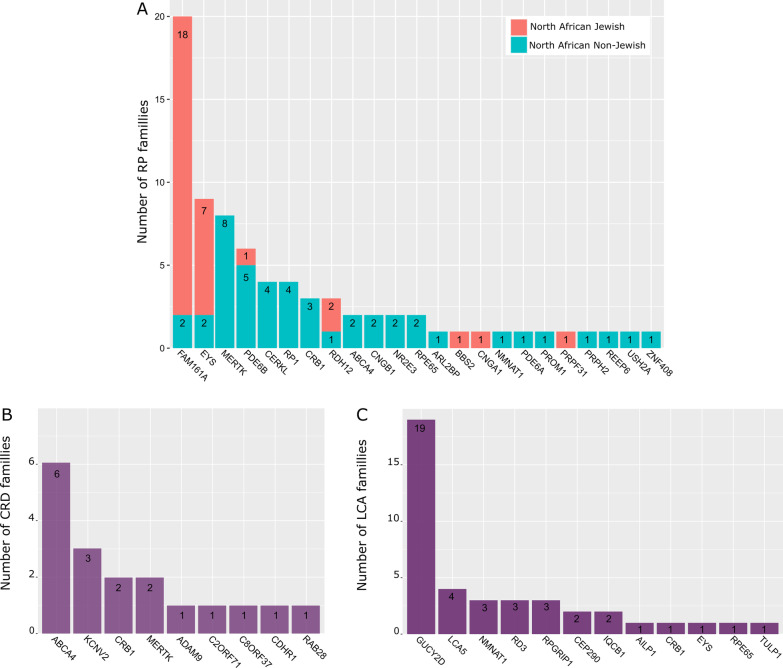
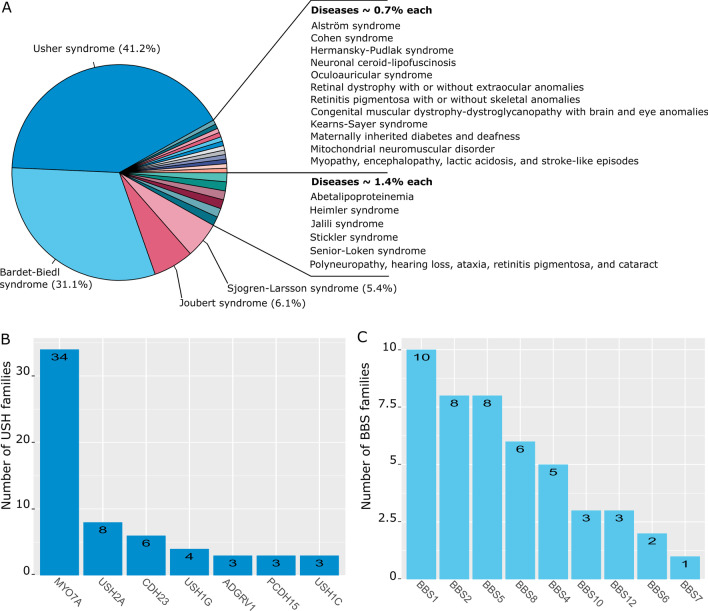
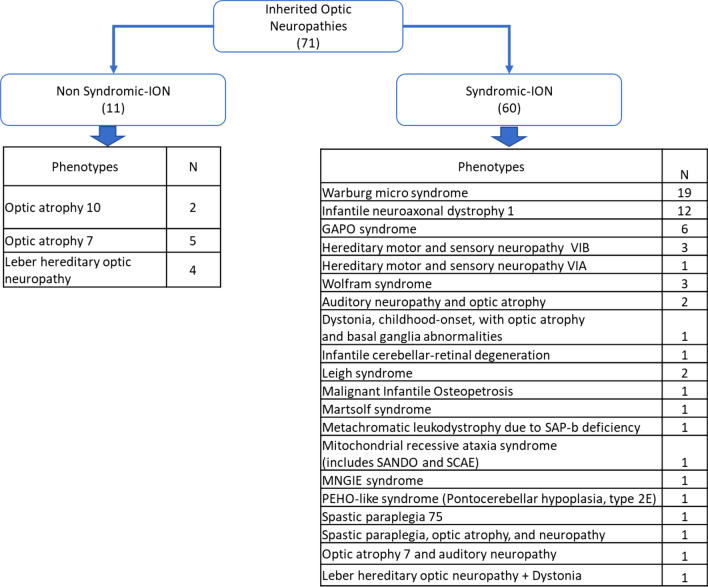
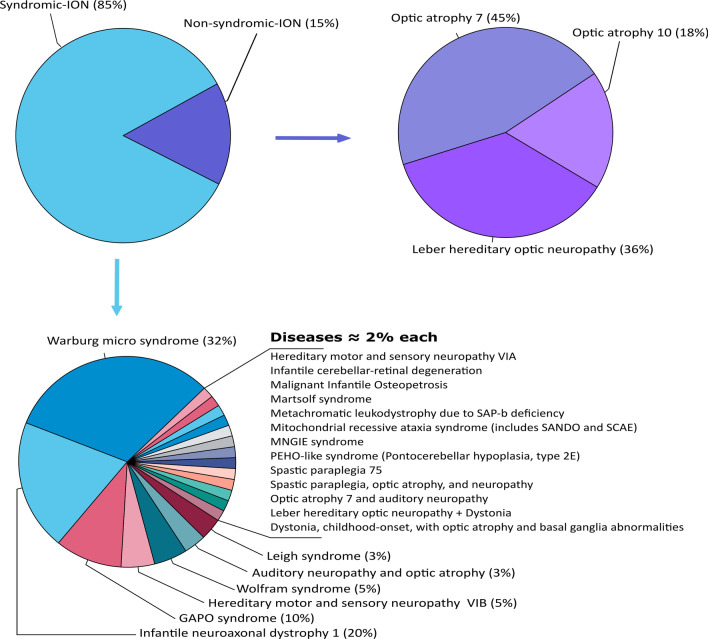
Main body: We compiled data from 413 families with IRD or ION together with their available molecular diagnosis. The proportion of IRD represents 82.8% of index cases, while ION accounted for 17.8%. Non-syndromic IRD were more frequent than syndromic ones, with photoreceptor alterations being the main cause of non-syndromic IRD, represented by retinitis pigmentosa, Leber congenital amaurosis, and cone-rod dystrophies, while ciliopathies constitute the major part of syndromic-IRD, in which the Usher and Bardet Biedl syndromes occupy 41.2% and 31.1%, respectively. We identified 71 ION families, 84.5% with a syndromic presentation, while surprisingly, non-syndromic ION are scarcely reported, with only 11 families with autosomal recessive optic atrophies related to OPA7 and OPA10 variants, or with the mitochondrial related Leber ION. Overall, consanguinity is a major cause of these diseases within North African countries, as 76.1% of IRD and 78.8% of ION investigated families were consanguineous, explaining the high rate of autosomal recessive inheritance pattern compared to the dominant one. In addition, we identified many founder mutations in small endogamous communities.
Short conclusion: As both IRD and ION diseases constitute a real public health burden, their under-diagnosis in North Africa due to the absence of physicians trained to the identification of inherited ophthalmologic presentations, together with the scarcity of tools for the molecular diagnosis represent major political, economic and health challenges for the future, to first establish accurate clinical diagnoses and then treat patients with the emergent therapies.
Keywords: Consanguinity; Genetic spectrum; Inherited optic neuropathies; Inherited retinal dystrophies; Molecular diagnosis; North Africa; Phenotypic spectrum.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interest.
Figures
Similar articles
-
Relative frequencies of inherited retinal dystrophies and optic neuropathies in Southern France: assessment of 21-year data management.Ophthalmic Epidemiol. 2013;20(1):13-25. doi: 10.3109/09286586.2012.737890. Ophthalmic Epidemiol. 2013. PMID: 23350551
-
Syndromic forms of inherited retinal dystrophies: a comprehensive molecular diagnosis of consanguineous Pakistani families using capture panel sequencing.Mol Vis. 2025 Mar 26;31:69-83. eCollection 2025. Mol Vis. 2025. PMID: 40384762 Free PMC article.
-
A multidisciplinary approach to inherited retinal dystrophies from diagnosis to initial care: a narrative review with inputs from clinical practice.Orphanet J Rare Dis. 2023 Jul 31;18(1):223. doi: 10.1186/s13023-023-02798-z. Orphanet J Rare Dis. 2023. PMID: 37525225 Free PMC article. Review.
-
Clinical and Molecular Features of a Chinese Cohort With Syndromic and Nonsyndromic Retinal Dystrophies Related to the CEP290 Gene.Am J Ophthalmol. 2023 Apr;248:96-106. doi: 10.1016/j.ajo.2022.11.023. Epub 2022 Dec 7. Am J Ophthalmol. 2023. PMID: 36493848
-
Gene Therapy in Hereditary Retinal Dystrophies: The Usefulness of Diagnostic Tools in Candidate Patient Selections.Int J Mol Sci. 2023 Sep 6;24(18):13756. doi: 10.3390/ijms241813756. Int J Mol Sci. 2023. PMID: 37762059 Free PMC article. Review.
Cited by
-
Retinal Organoids from Induced Pluripotent Stem Cells of Patients with Inherited Retinal Diseases: A Systematic Review.Stem Cell Rev Rep. 2025 Jan;21(1):167-197. doi: 10.1007/s12015-024-10802-7. Epub 2024 Oct 18. Stem Cell Rev Rep. 2025. PMID: 39422807 Free PMC article.
-
Bardet-Biedl Syndrome: Current Perspectives and Clinical Outlook.Ther Clin Risk Manag. 2023 Jan 30;19:115-132. doi: 10.2147/TCRM.S338653. eCollection 2023. Ther Clin Risk Manag. 2023. PMID: 36741589 Free PMC article. Review.
-
Characterizing the Genetic Basis for Inherited Retinal Disease: Lessons Learned From the Foundation Fighting Blindness Clinical Consortium's Gene Poll.Invest Ophthalmol Vis Sci. 2025 Feb 3;66(2):12. doi: 10.1167/iovs.66.2.12. Invest Ophthalmol Vis Sci. 2025. PMID: 39908130 Free PMC article.
-
Identification and functional characterization of ABCA4 gene variants in three patients with Stargardt disease or retinitis pigmentosa.Front Genet. 2025 Jun 18;16:1516872. doi: 10.3389/fgene.2025.1516872. eCollection 2025. Front Genet. 2025. PMID: 40606666 Free PMC article.
-
RetiGene, a comprehensive gene atlas for inherited retinal diseases (IRDs).bioRxiv [Preprint]. 2025 Jun 8:2025.06.08.653722. doi: 10.1101/2025.06.08.653722. bioRxiv. 2025. PMID: 40661613 Free PMC article. Preprint.
References
-
- Worldometers, African Countries by population. 2021. https://www.worldometers.info/population/countries-in-africa-by-population/
-
- Newman JL. The peopling of Africa: a geographic interpretation. New Haven: Yale University Press; 1995. p. 235.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
