

Endocrine Manifestations and New Developments in Mitochondrial Disease
- PMID: 35552684
- PMCID: PMC9113134
- DOI: 10.1210/endrev/bnab036
Endocrine Manifestations and New Developments in Mitochondrial Disease
Abstract
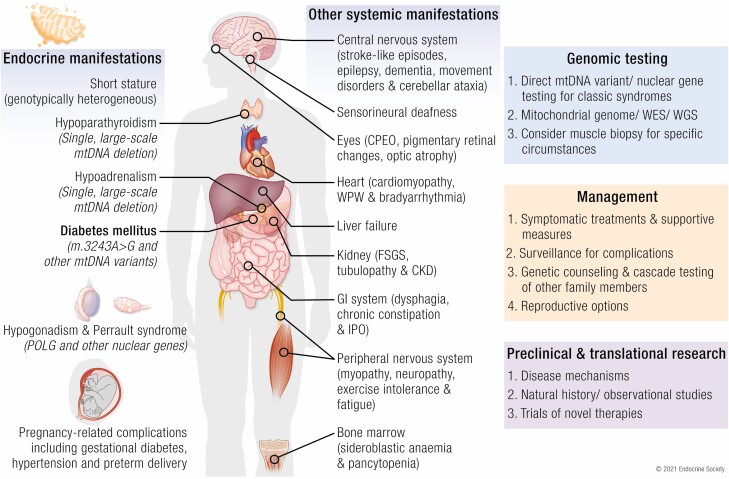
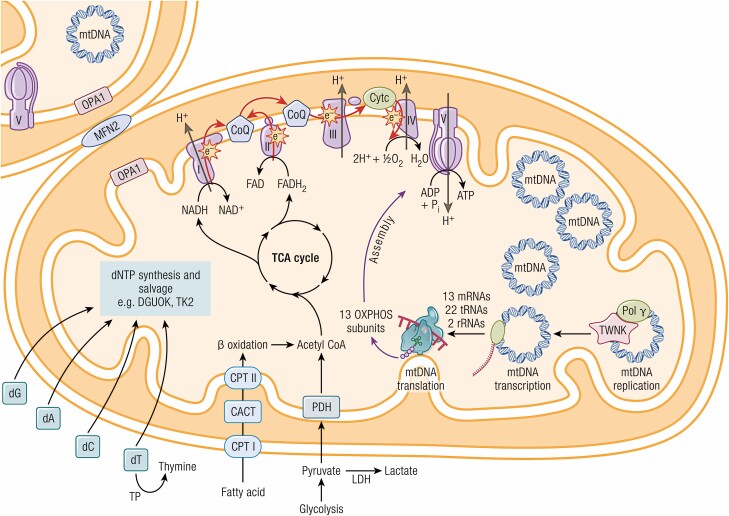
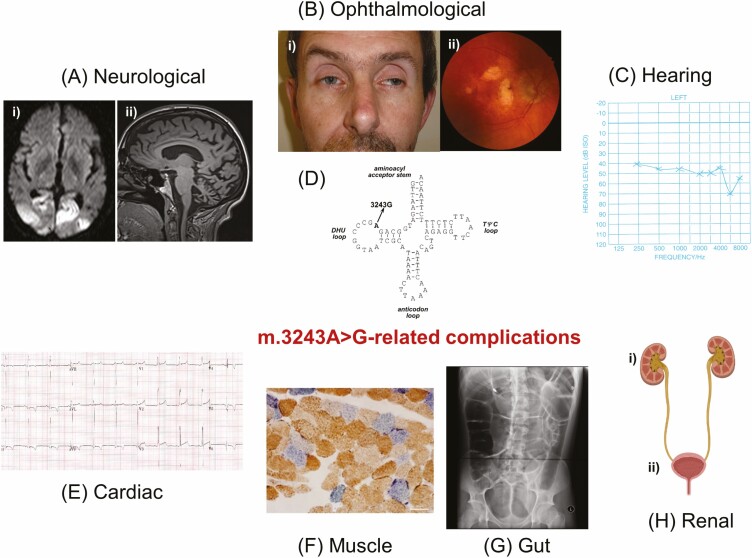
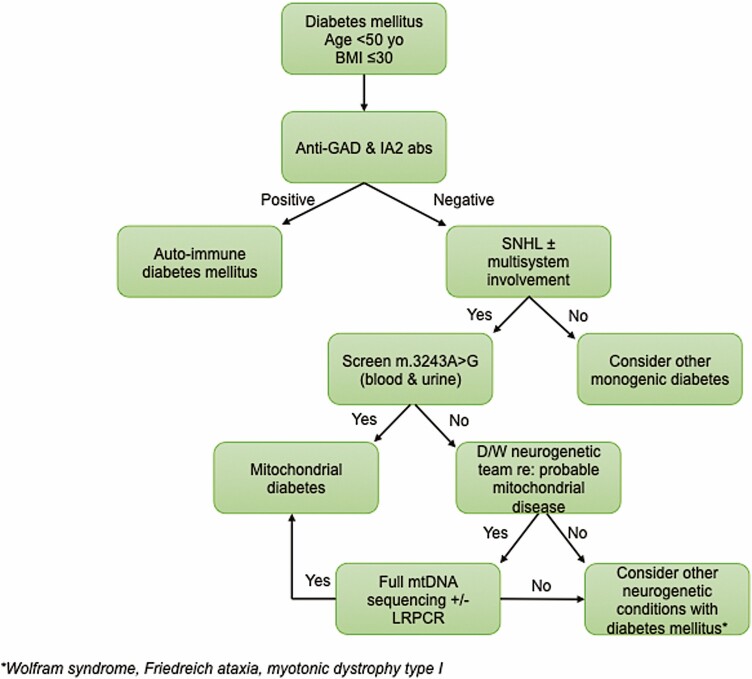
Mitochondrial diseases are a group of common inherited diseases causing disruption of oxidative phosphorylation. Some patients with mitochondrial disease have endocrine manifestations, with diabetes mellitus being predominant but also include hypogonadism, hypoadrenalism, and hypoparathyroidism. There have been major developments in mitochondrial disease over the past decade that have major implications for all patients. The collection of large cohorts of patients has better defined the phenotype of mitochondrial diseases and the majority of patients with endocrine abnormalities have involvement of several other systems. This means that patients with mitochondrial disease and endocrine manifestations need specialist follow-up because some of the other manifestations, such as stroke-like episodes and cardiomyopathy, are potentially life threatening. Also, the development and follow-up of large cohorts of patients means that there are clinical guidelines for the management of patients with mitochondrial disease. There is also considerable research activity to identify novel therapies for the treatment of mitochondrial disease. The revolution in genetics, with the introduction of next-generation sequencing, has made genetic testing more available and establishing a precise genetic diagnosis is important because it will affect the risk for involvement for different organ systems. Establishing a genetic diagnosis is also crucial because important reproductive options have been developed that will prevent the transmission of mitochondrial disease because of mitochondrial DNA variants to the next generation.
Keywords: MIDD; clinical management; diabetes mellitus; genomic testing; mitochondrial DNA; reproductive options.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Endocrine Society.
Figures
Similar articles
-
Mitochondrial disease and endocrine dysfunction.Nat Rev Endocrinol. 2017 Feb;13(2):92-104. doi: 10.1038/nrendo.2016.151. Epub 2016 Oct 7. Nat Rev Endocrinol. 2017. PMID: 27716753 Review.
-
[Mitochondrial diabetes: clinical features, diagnosis and management].Rev Med Interne. 2010 Mar;31(3):216-21. doi: 10.1016/j.revmed.2008.11.017. Epub 2009 Mar 18. Rev Med Interne. 2010. PMID: 19299044 Review. French.
-
Mitochondrial diseases.Nat Rev Dis Primers. 2016 Oct 20;2:16080. doi: 10.1038/nrdp.2016.80. Nat Rev Dis Primers. 2016. PMID: 27775730 Review.
-
The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation--implications for diagnosis and management.J Neurol Neurosurg Psychiatry. 2013 Aug;84(8):936-8. doi: 10.1136/jnnp-2012-303528. Epub 2013 Jan 25. J Neurol Neurosurg Psychiatry. 2013. PMID: 23355809
-
Endocrine disorders in mitochondrial disease.Mol Cell Endocrinol. 2013 Oct 15;379(1-2):2-11. doi: 10.1016/j.mce.2013.06.004. Epub 2013 Jun 13. Mol Cell Endocrinol. 2013. PMID: 23769710 Free PMC article. Review.
Cited by
-
Neonatal and Syndromic Forms of Diabetes.Curr Diab Rep. 2025 Mar 25;25(1):26. doi: 10.1007/s11892-024-01567-x. Curr Diab Rep. 2025. PMID: 40128490 Free PMC article. Review.
-
Endocrine Dysfunction in Primary Mitochondrial Diseases.Endocr Rev. 2025 May 9;46(3):376-396. doi: 10.1210/endrev/bnaf002. Endocr Rev. 2025. PMID: 39891580 Free PMC article. Review.
-
Diabetes, macrocytosis, and skin changes in large-scale mtDNA deletion.J Pediatr Endocrinol Metab. 2025 Mar 10;38(6):663-667. doi: 10.1515/jpem-2025-0016. Print 2025 Jun 26. J Pediatr Endocrinol Metab. 2025. PMID: 40055929
-
Penetrance and expressivity of mitochondrial variants in a large clinically unselected population.Hum Mol Genet. 2024 Feb 18;33(5):465-474. doi: 10.1093/hmg/ddad194. Hum Mol Genet. 2024. PMID: 37988592 Free PMC article.
-
Polycystic Ovary Syndrome and Oxidative Stress-From Bench to Bedside.Int J Mol Sci. 2023 Sep 15;24(18):14126. doi: 10.3390/ijms241814126. Int J Mol Sci. 2023. PMID: 37762427 Free PMC article. Review.
References
-
- Ng YS, Bindoff LA, Gorman GS, et al. Mitochondrial disease in adults: recent advances and future promise. Lancet Neurol. 2021;20(7):573-584. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
