

Clinical Study of 30 Novel KCNQ2 Variants/Deletions in KCNQ2-Related Disorders
- PMID: 35557555
- PMCID: PMC9088225
- DOI: 10.3389/fnmol.2022.809810
Clinical Study of 30 Novel KCNQ2 Variants/Deletions in KCNQ2-Related Disorders
Abstract
Background: KCNQ2-related disorder is typically characterized as neonatal onset seizure and epileptic encephalopathy. The relationship between its phenotype and genotype is still elusive. This study aims to provide clinical features, management, and prognosis of patients with novel candidate variants of the KCNQ2 gene.
Methods: We enrolled patients with novel variants in the KCNQ2 gene from the China Neonatal Genomes Project between January 2018 and January 2021. All patients underwent next-generation sequencing tests and genetic data were analyzed by an in-house pipeline. The pathogenicity of variants was classified according to the guideline of the American College of Medical Genetics. Each case was evaluated by two geneticists back to back. Patients' information was acquired from clinical records.
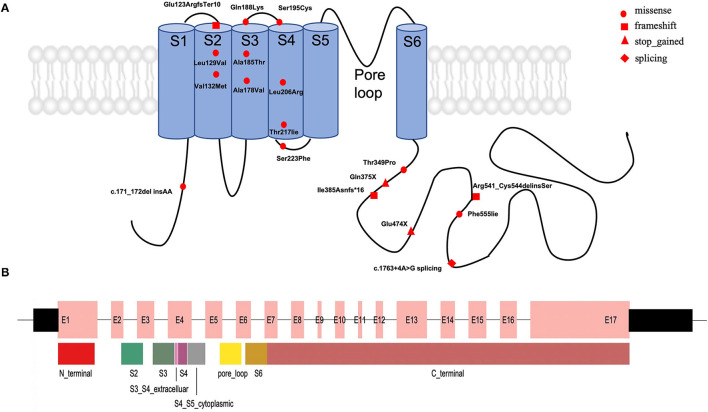
Results: A total of 30 unrelated patients with novel variants in the KCNQ2 gene were identified, including 19 patients with single-nucleotide variants (SNVs) and 11 patients with copy number variants (CNVs). For the 19 SNVs, 12 missense variants and 7 truncating variants were identified. Of them, 36.8% (7/19) of the KCNQ2 variants were located in C-terminal regions, 15.7% (3/19) in segment S2, and 15.7% (3/19) in segment S4. Among them, 18 of 19 patients experienced seizures in the early neonatal period. However, one patient presented neurodevelopmental delay (NDD) as initial phenotype when he was 2 months old, and he had severe NDD when he was 3 years old. This patient did not present seizure but had abnormal electrographic background activity and brain imaging. Moreover, for the 11 patients with CNVs, 20q13.3 deletions involving EEF1A2, KCNQ2, and CHRNA4 genes were detected. All of them presented neonatal-onset seizures, responded to antiepileptic drugs, and had normal neurological development.
Conclusion: In this study, patients with novel KCNQ2 variants have variable phenotypes, whereas patients with 20q13.3 deletion involving EEF1A2, KCNQ2, and CHRNA4 genes tend to have normal neurological development.
Keywords: KCNQ2; Kv7.2; epilepsy; epileptic encephalopathy; newborn.
Copyright © 2022 Xiao, Chen, Xu, Chen, Dong, Yang, Wu, Chen, Li, Zhuang, Chen, Zhou, Wang and Zhou.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Contiguous deletion of KCNQ2 and CHRNA4 may cause a different disorder from benign familial neonatal seizures.Epilepsy Behav Case Rep. 2013 Mar 1;1:35-8. doi: 10.1016/j.ebcr.2013.01.004. eCollection 2013. Epilepsy Behav Case Rep. 2013. PMID: 25667822 Free PMC article.
-
KCNQ2 R144 variants cause neurodevelopmental disability with language impairment and autistic features without neonatal seizures through a gain-of-function mechanism.EBioMedicine. 2022 Jul;81:104130. doi: 10.1016/j.ebiom.2022.104130. Epub 2022 Jun 30. EBioMedicine. 2022. PMID: 35780567 Free PMC article.
-
KCNQ2 mutations in childhood nonlesional epilepsy: Variable phenotypes and a novel mutation in a case series.Mol Genet Genomic Med. 2019 Jul;7(7):e00816. doi: 10.1002/mgg3.816. Epub 2019 Jun 14. Mol Genet Genomic Med. 2019. PMID: 31199083 Free PMC article.
-
Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant.Epilepsia. 2017 Jan;58(1):e10-e15. doi: 10.1111/epi.13601. Epub 2016 Nov 9. Epilepsia. 2017. PMID: 27861786 Free PMC article.
-
The Role of Kv7.2 in Neurodevelopment: Insights and Gaps in Our Understanding.Front Physiol. 2020 Oct 28;11:570588. doi: 10.3389/fphys.2020.570588. eCollection 2020. Front Physiol. 2020. PMID: 33192566 Free PMC article. Review.
Cited by
-
Biophysical and structural mechanisms of epilepsy-associated mutations in the S4-S5 Linker of KCNQ2 channels.Channels (Austin). 2025 Dec;19(1):2464735. doi: 10.1080/19336950.2025.2464735. Epub 2025 Feb 19. Channels (Austin). 2025. PMID: 39971736 Free PMC article.
-
Novel KCNQ2 Variants Related to a Variable Phenotypic Spectrum Ranging from Epilepsy with Auditory Features to Severe Developmental and Epileptic Encephalopathies.Int J Mol Sci. 2024 Dec 31;26(1):295. doi: 10.3390/ijms26010295. Int J Mol Sci. 2024. PMID: 39796146 Free PMC article.
References
-
- Abidi A., Devaux J. J., Molinari F., Alcaraz G., Michon F. X., Sutera-Sardo J., et al. . (2015). A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol Dis. 80, 80–92. 10.1016/j.nbd.2015.04.017 - DOI - PubMed
-
- Béna F., Bottani A., Marcelli F., Sizonenko L. D., Conrad B., Dahoun S. (2007). A de novo 1.1-1.6 Mb subtelomeric deletion of chromosome 20q13.33 in a patient with learning difficulties but without obvious dysmorphic features. Am. J. Med. Genet. A. 143a, 1894–1899. 10.1002/ajmg.a.31789 - DOI - PubMed
LinkOut - more resources
Full Text Sources