

Myocardial injury, troponin release, and cardiomyocyte death in brief ischemia, failure, and ventricular remodeling
- PMID: 35559722
- PMCID: PMC9169835
- DOI: 10.1152/ajpheart.00093.2022
Myocardial injury, troponin release, and cardiomyocyte death in brief ischemia, failure, and ventricular remodeling
Abstract
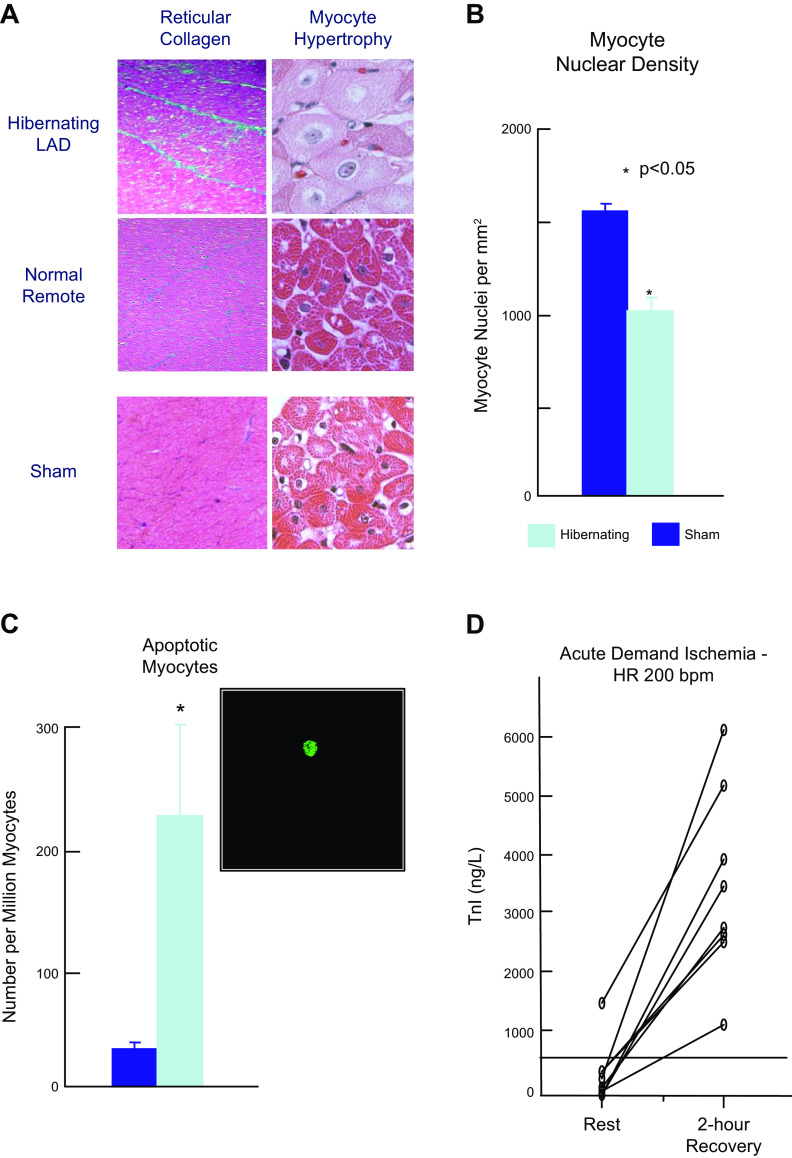
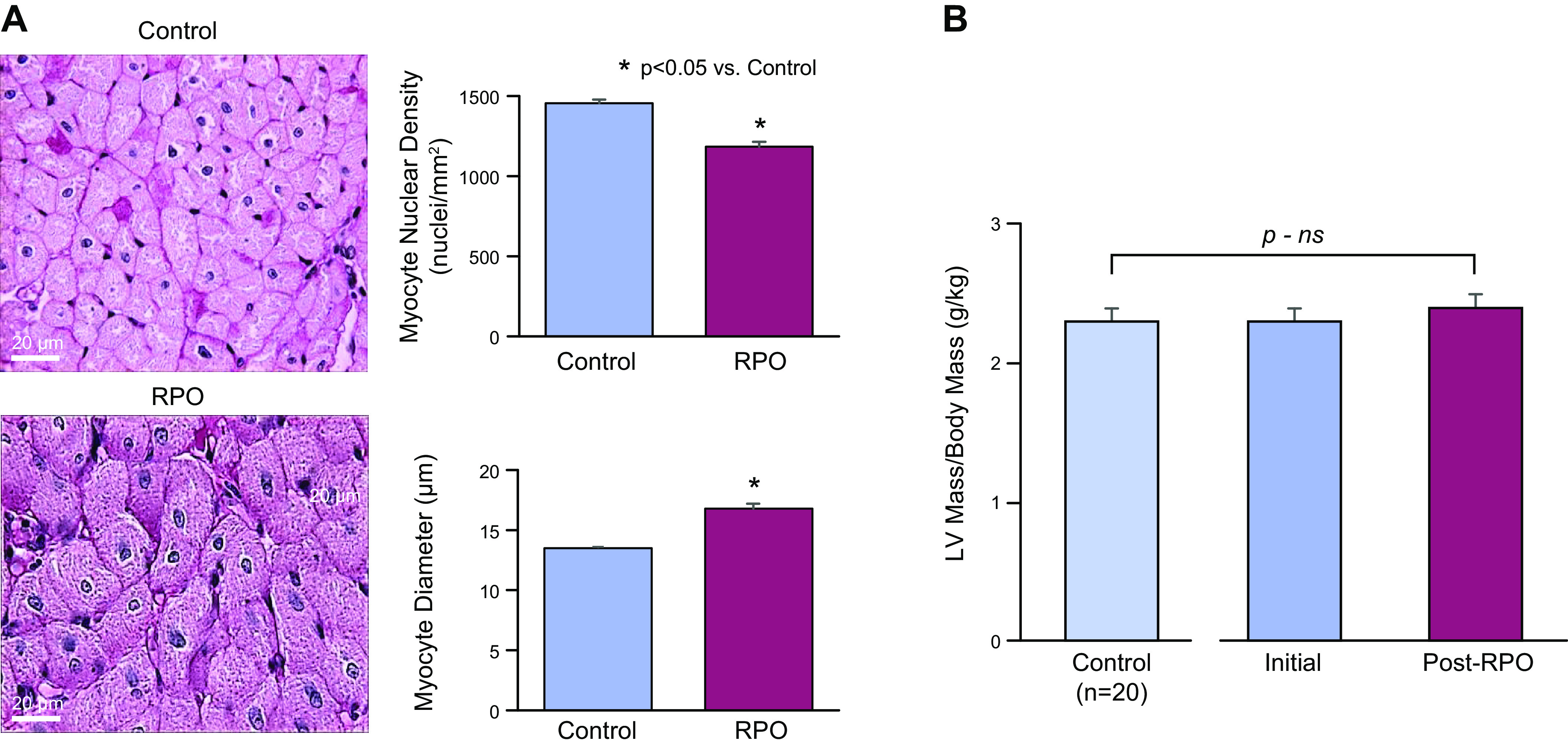
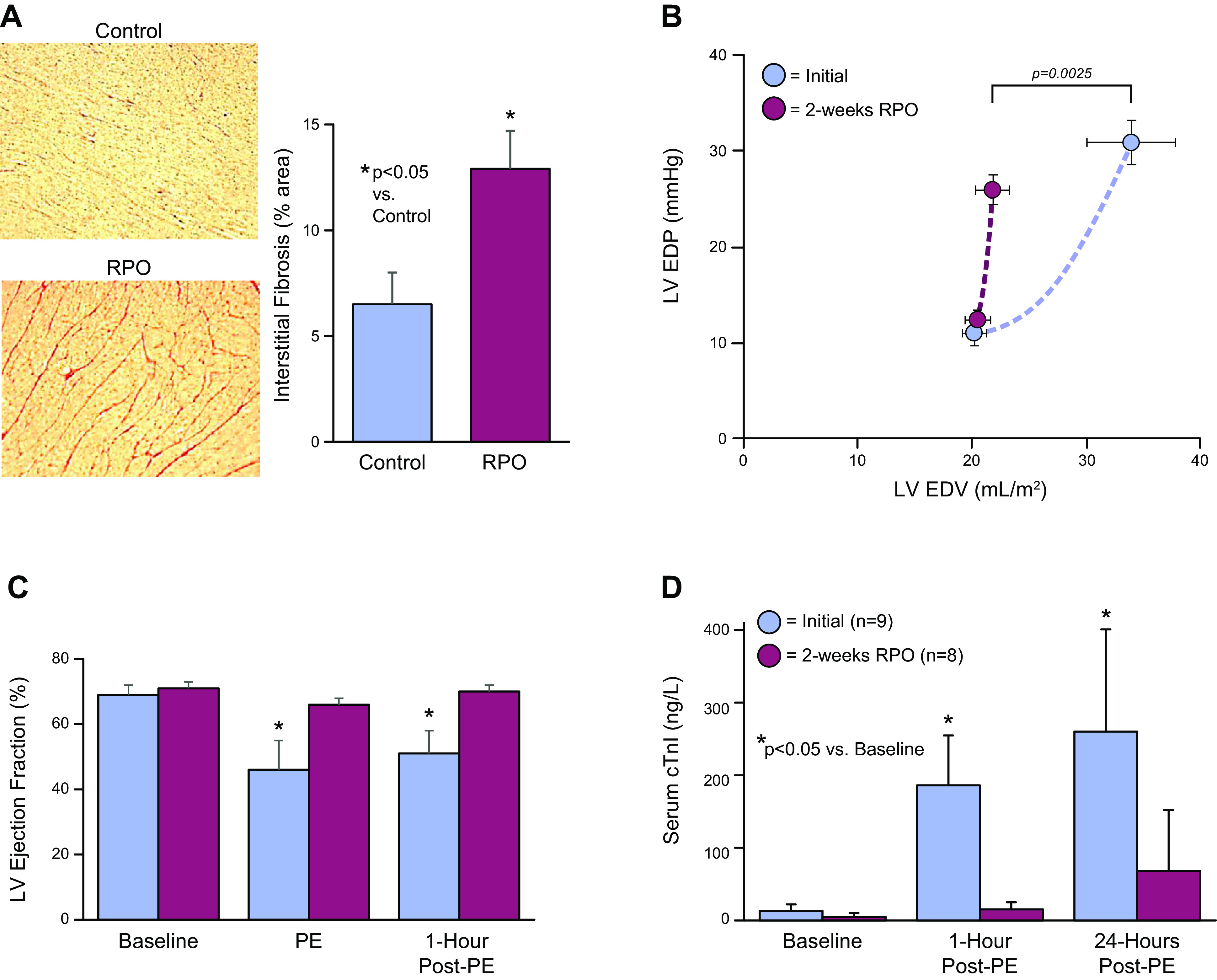
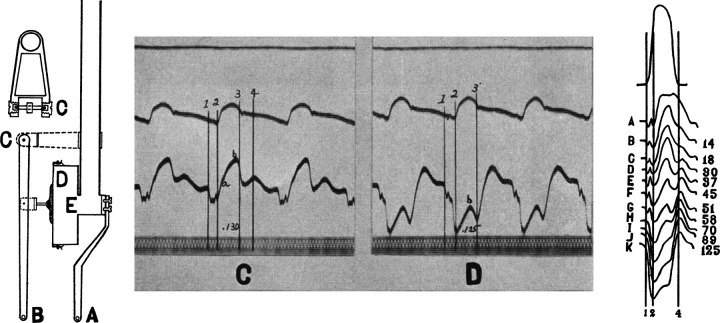
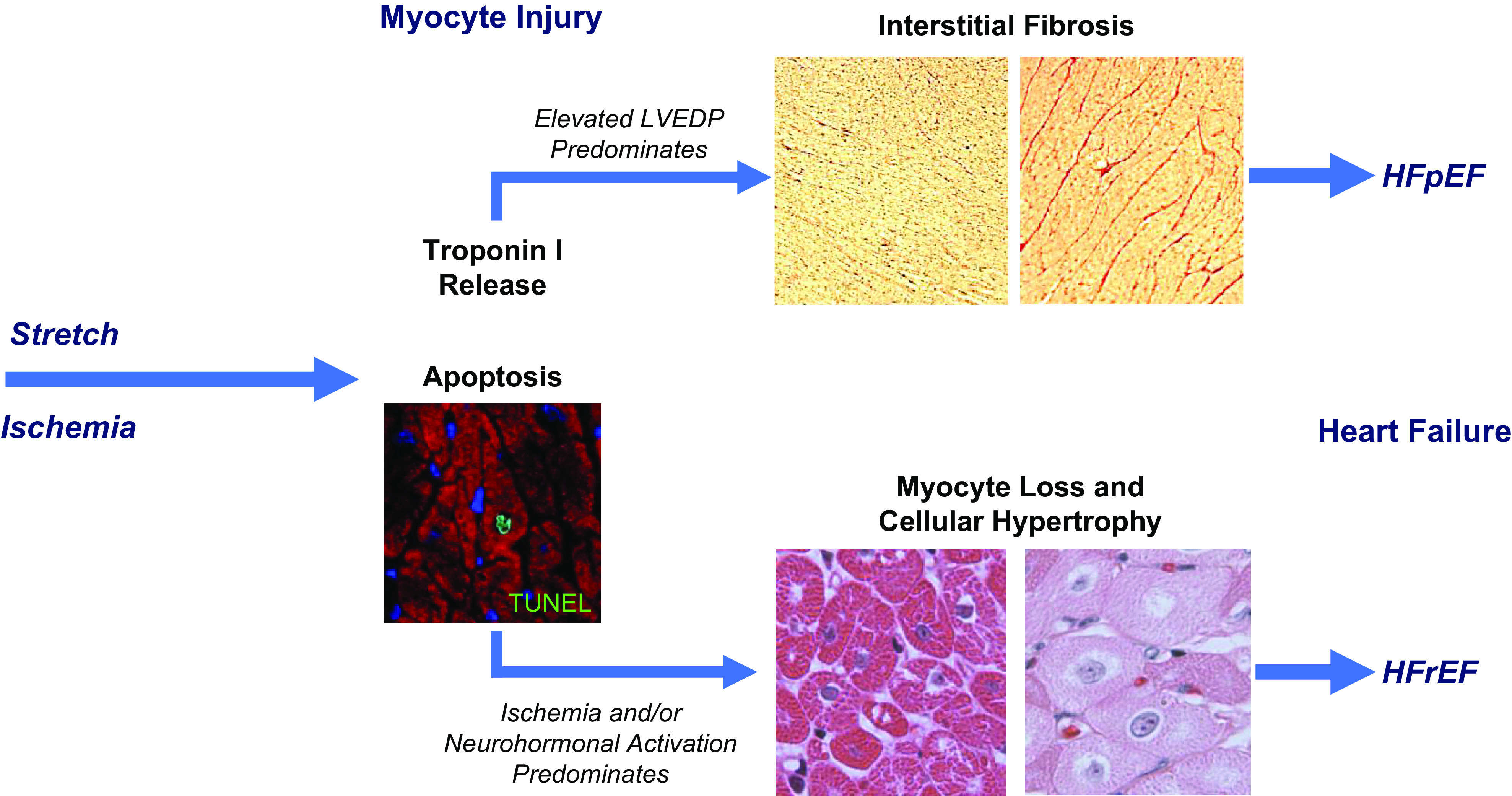
Troponin released from irreversibly injured myocytes is the gold standard biomarker for the rapid identification of an acute coronary syndrome. In acute myocardial infarction, necrotic cell death is characterized by sarcolemmal disruption in response to a critical level of energy depletion after more than 15 min of ischemia. Although troponin I and T are highly specific for cardiomyocyte death, high-sensitivity assays have demonstrated that measurable circulating levels of troponin are present in many normal subjects. In addition, transient as well as chronic elevations have been demonstrated in many disease states not clearly associated with myocardial ischemia. The latter observations have given rise to the clinical concept of myocardial injury. This review will summarize evidence supporting the notion that circulating troponin levels parallel the extent of myocyte apoptosis in normal ventricular remodeling and in pathophysiological conditions not associated with infarction or necrosis. It will review the evidence that myocyte apoptosis can be accelerated by diastolic strain from elevated ventricular preload and systolic strain from dyskinesis after brief episodes of ischemia too short to cause a critical level of myocyte energy depletion. We then show how chronic, low rates of myocyte apoptosis from endogenous myocyte turnover, repetitive ischemia, or repetitive elevations in left ventricular diastolic pressure can lead to significant myocyte loss in the absence of neurohormonal stimulation. Finally, we posit that the differential response to strain-induced injury in heart failure may determine whether progressive myocyte loss and heart failure with reduced ejection fraction or interstitial fibrosis and heart failure with preserved ejection fraction become the heart failure phenotype.
Keywords: HFpEF; HFrEF; apoptosis; ischemia; troponin.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the author.
Figures
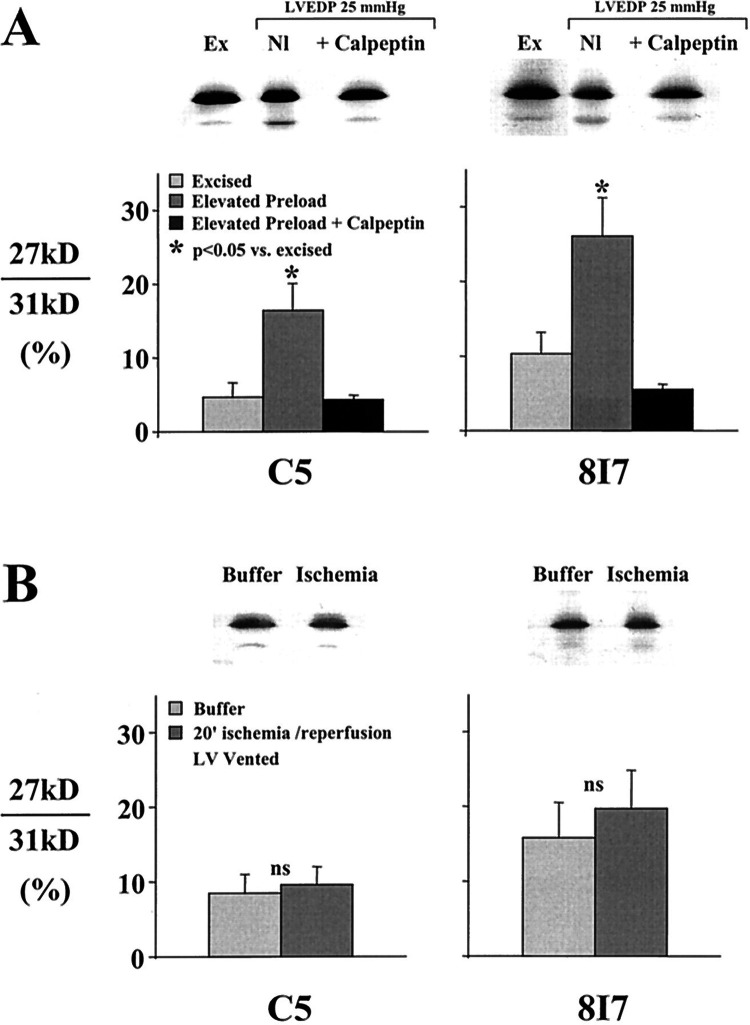
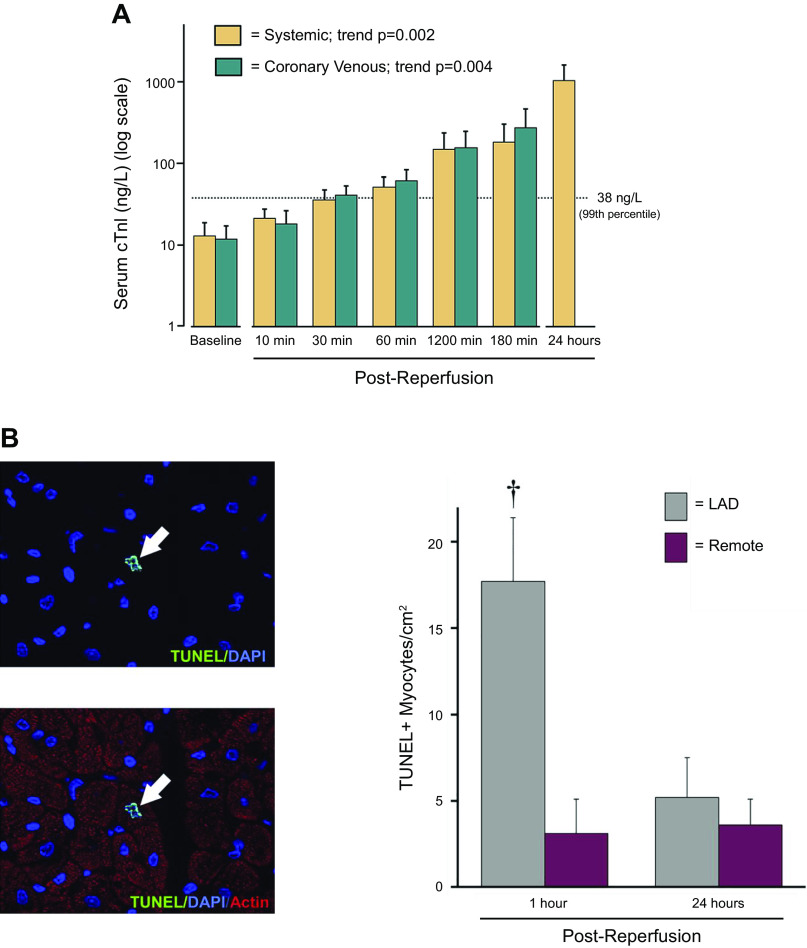
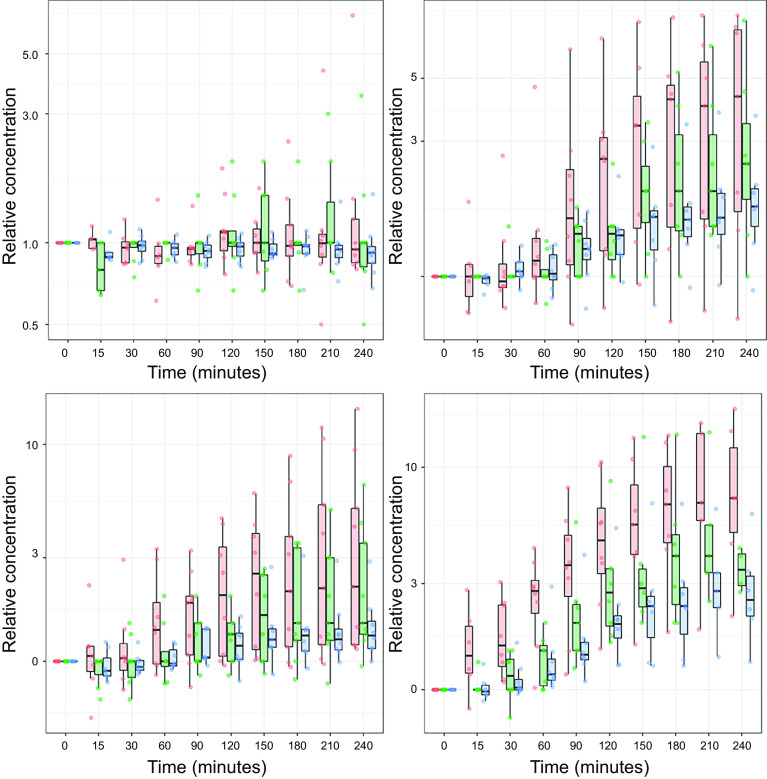
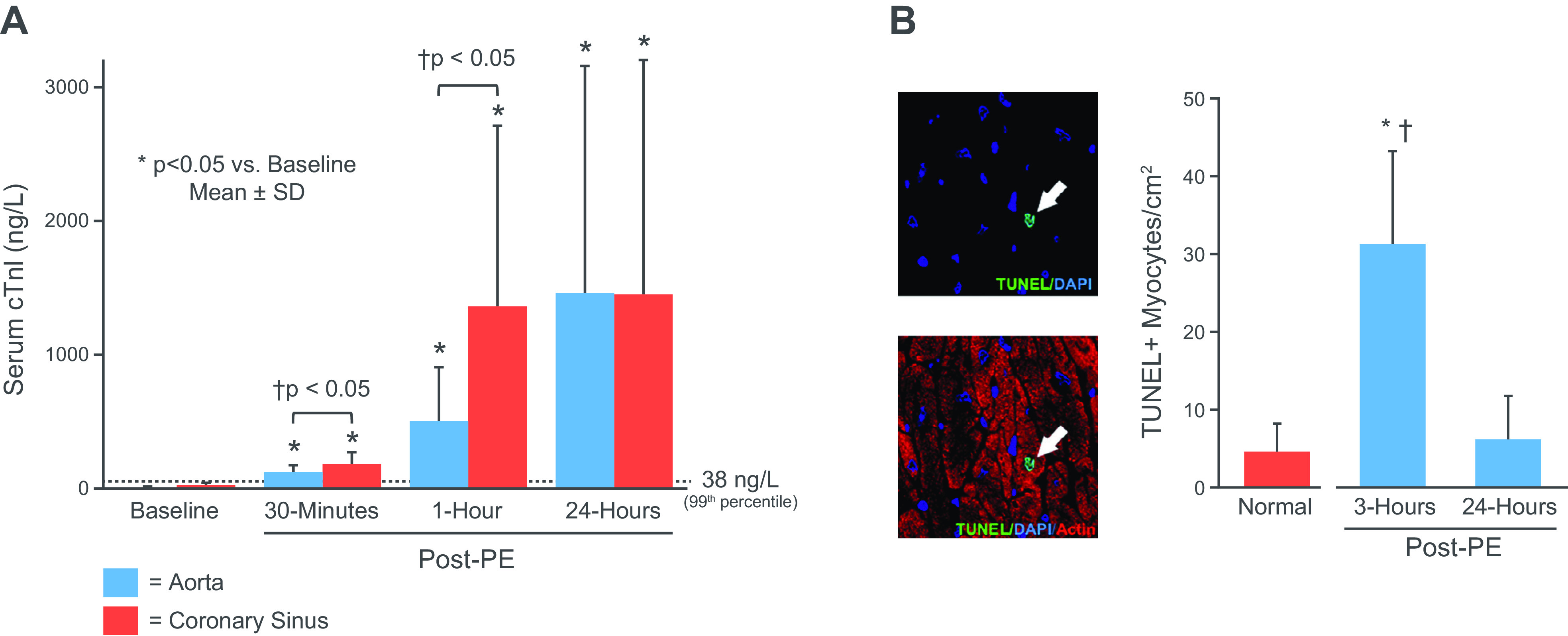
References
-
- Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E, Anversa P. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol 28: 2005–2016, 1996. doi: 10.1006/jmcc.1996.0193. - DOI - PubMed
-
- Jennings RB, Reimer KA, Hill ML, Mayer SE. Total ischemia in dog hearts, in vitro 1. Comparison of high energy phosphate production, utilization, and depletion, and of adenine nucleotide catabolism in total ischemia in vitro vs. severe ischemia in vivo. Circ Res 49: 892–900, 1981. doi: 10.1161/01.res.49.4.892. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical