

TopHap: rapid inference of key phylogenetic structures from common haplotypes in large genome collections with limited diversity
- PMID: 35561179
- PMCID: PMC9113349
- DOI: 10.1093/bioinformatics/btac186
TopHap: rapid inference of key phylogenetic structures from common haplotypes in large genome collections with limited diversity
Abstract
Motivation: Building reliable phylogenies from very large collections of sequences with a limited number of phylogenetically informative sites is challenging because sequencing errors and recurrent/backward mutations interfere with the phylogenetic signal, confounding true evolutionary relationships. Massive global efforts of sequencing genomes and reconstructing the phylogeny of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) strains exemplify these difficulties since there are only hundreds of phylogenetically informative sites but millions of genomes. For such datasets, we set out to develop a method for building the phylogenetic tree of genomic haplotypes consisting of positions harboring common variants to improve the signal-to-noise ratio for more accurate and fast phylogenetic inference of resolvable phylogenetic features.
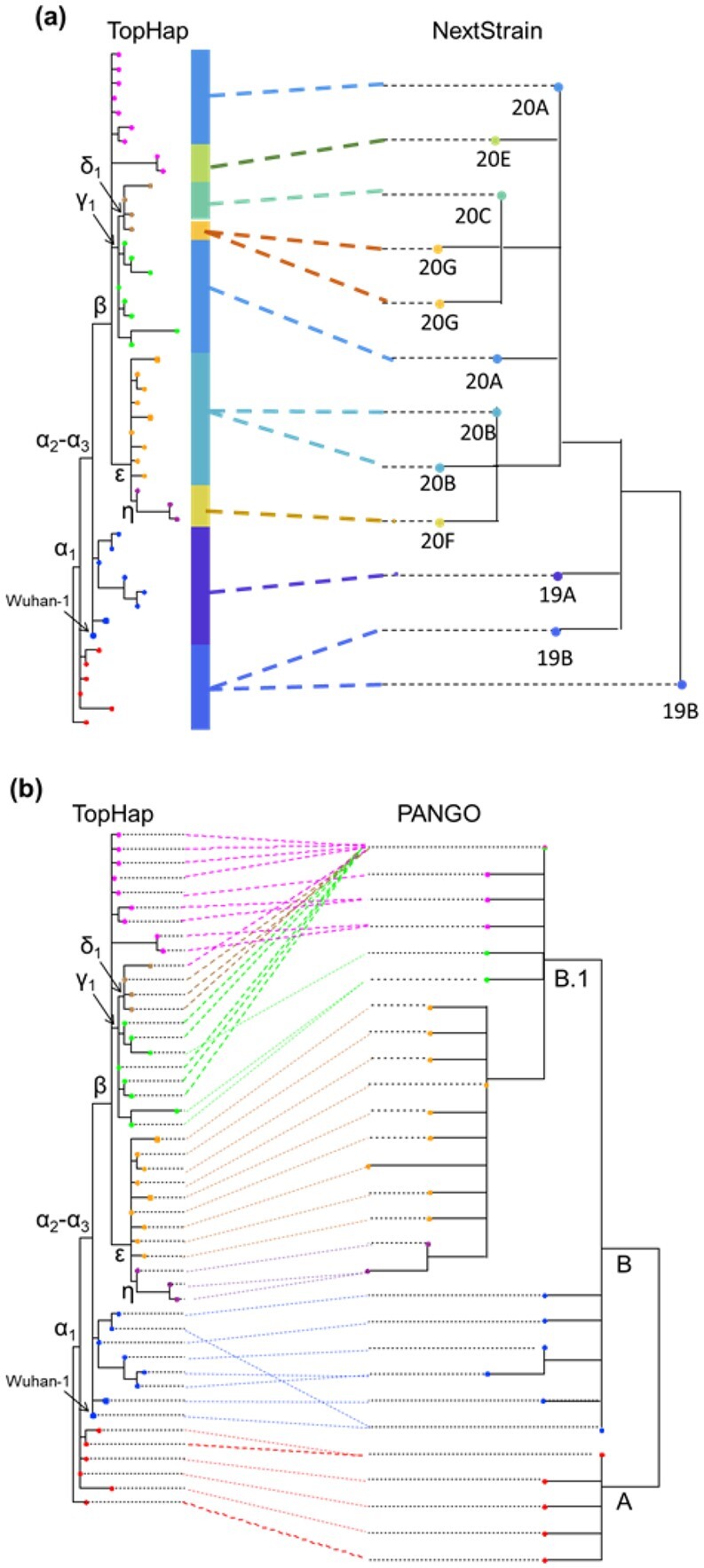
Results: We present the TopHap approach that determines spatiotemporally common haplotypes of common variants and builds their phylogeny at a fraction of the computational time of traditional methods. We develop a bootstrap strategy that resamples genomes spatiotemporally to assess topological robustness. The application of TopHap to build a phylogeny of 68 057 SARS-CoV-2 genomes (68KG) from the first year of the pandemic produced an evolutionary tree of major SARS-CoV-2 haplotypes. This phylogeny is concordant with the mutation tree inferred using the co-occurrence pattern of mutations and recovers key phylogenetic relationships from more traditional analyses. We also evaluated alternative roots of the SARS-CoV-2 phylogeny and found that the earliest sampled genomes in 2019 likely evolved by four mutations of the most recent common ancestor of all SARS-CoV-2 genomes. An application of TopHap to more than 1 million SARS-CoV-2 genomes reconstructed the most comprehensive evolutionary relationships of major variants, which confirmed the 68KG phylogeny and provided evolutionary origins of major and recent variants of concern.
Availability and implementation: TopHap is available at https://github.com/SayakaMiura/TopHap.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2022. Published by Oxford University Press.
Figures






References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
