

Analysing high-throughput sequencing data in Python with HTSeq 2.0
- PMID: 35561197
- PMCID: PMC9113351
- DOI: 10.1093/bioinformatics/btac166
Analysing high-throughput sequencing data in Python with HTSeq 2.0
Abstract
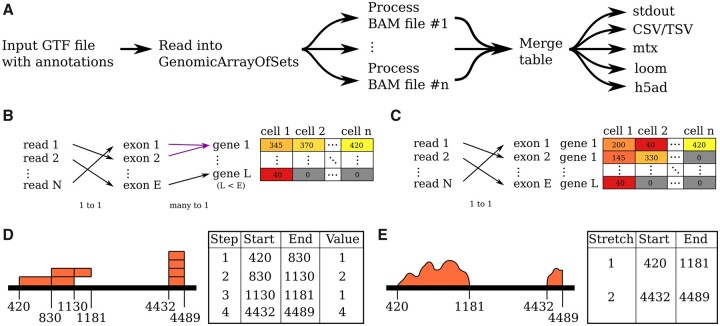
Summary: HTSeq 2.0 provides a more extensive application programming interface including a new representation for sparse genomic data, enhancements for htseq-count to suit single-cell omics, a new script for data using cell and molecular barcodes, improved documentation, testing and deployment, bug fixes and Python 3 support.
Availability and implementation: HTSeq 2.0 is released as an open-source software under the GNU General Public License and is available from the Python Package Index at https://pypi.python.org/pypi/HTSeq. The source code is available on Github at https://github.com/htseq/htseq.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2022. Published by Oxford University Press.
Figures
References
-
- Beazley D.M. (2003) Automated scientific software scripting with SWIG. Fut. Generat. Comput. Syst. FGCS, 19, 599–609.
-
- Behnel S. et al. (2011) Cython: the best of both worlds. Comput. Sci. Eng., 13, 31–39.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
