

FIRM: Flexible integration of single-cell RNA-sequencing data for large-scale multi-tissue cell atlas datasets
- PMID: 35561293
- PMCID: PMC9487597
- DOI: 10.1093/bib/bbac167
FIRM: Flexible integration of single-cell RNA-sequencing data for large-scale multi-tissue cell atlas datasets
Abstract
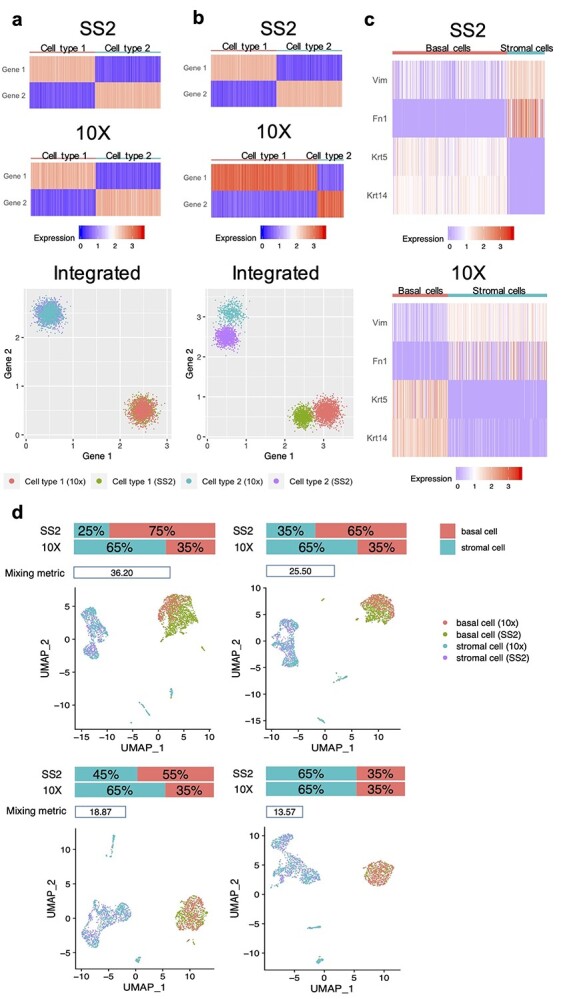
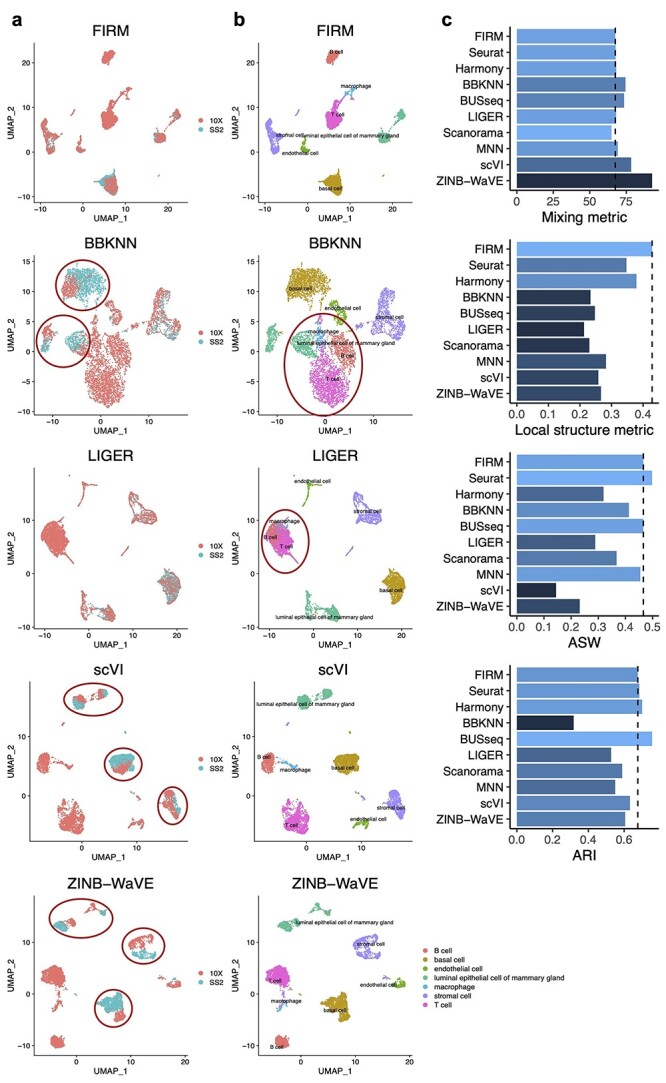
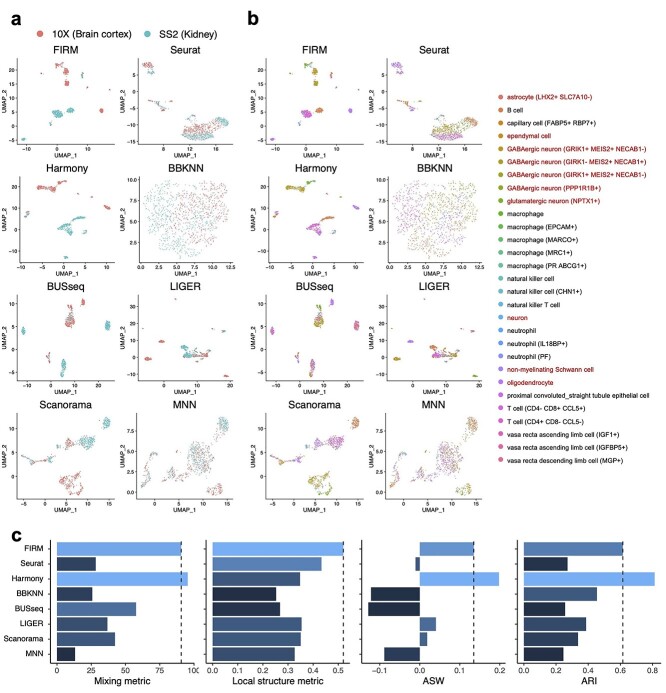
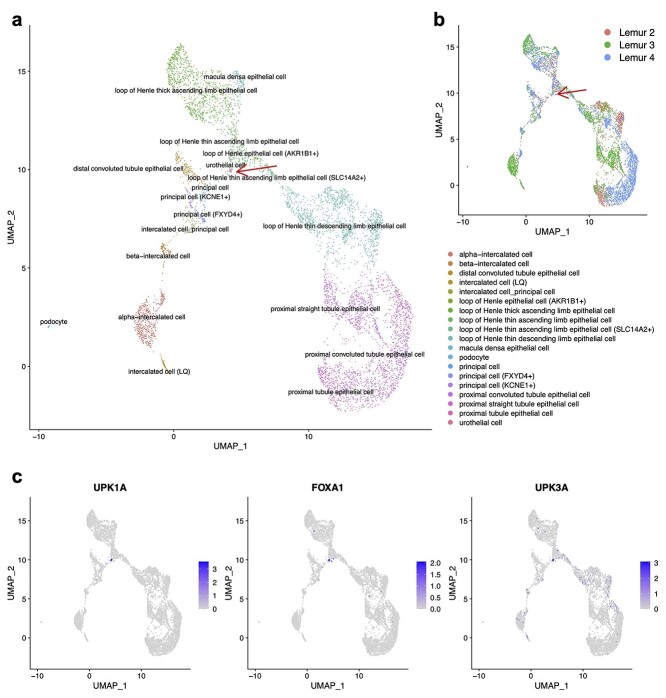
Single-cell RNA-sequencing (scRNA-seq) is being used extensively to measure the mRNA expression of individual cells from deconstructed tissues, organs and even entire organisms to generate cell atlas references, leading to discoveries of novel cell types and deeper insight into biological trajectories. These massive datasets are usually collected from many samples using different scRNA-seq technology platforms, including the popular SMART-Seq2 (SS2) and 10X platforms. Inherent heterogeneities between platforms, tissues and other batch effects make scRNA-seq data difficult to compare and integrate, especially in large-scale cell atlas efforts; yet, accurate integration is essential for gaining deeper insights into cell biology. We present FIRM, a re-scaling algorithm which accounts for the effects of cell type compositions, and achieve accurate integration of scRNA-seq datasets across multiple tissue types, platforms and experimental batches. Compared with existing state-of-the-art integration methods, FIRM provides accurate mixing of shared cell type identities and superior preservation of original structure without overcorrection, generating robust integrated datasets for downstream exploration and analysis. FIRM is also a facile way to transfer cell type labels and annotations from one dataset to another, making it a reliable and versatile tool for scRNA-seq analysis, especially for cell atlas data integration.
Keywords: bioinformatics; cell atlas; data integration; single-cell RNA sequencing.
© The Author(s) 2022. Published by Oxford University Press.
Figures
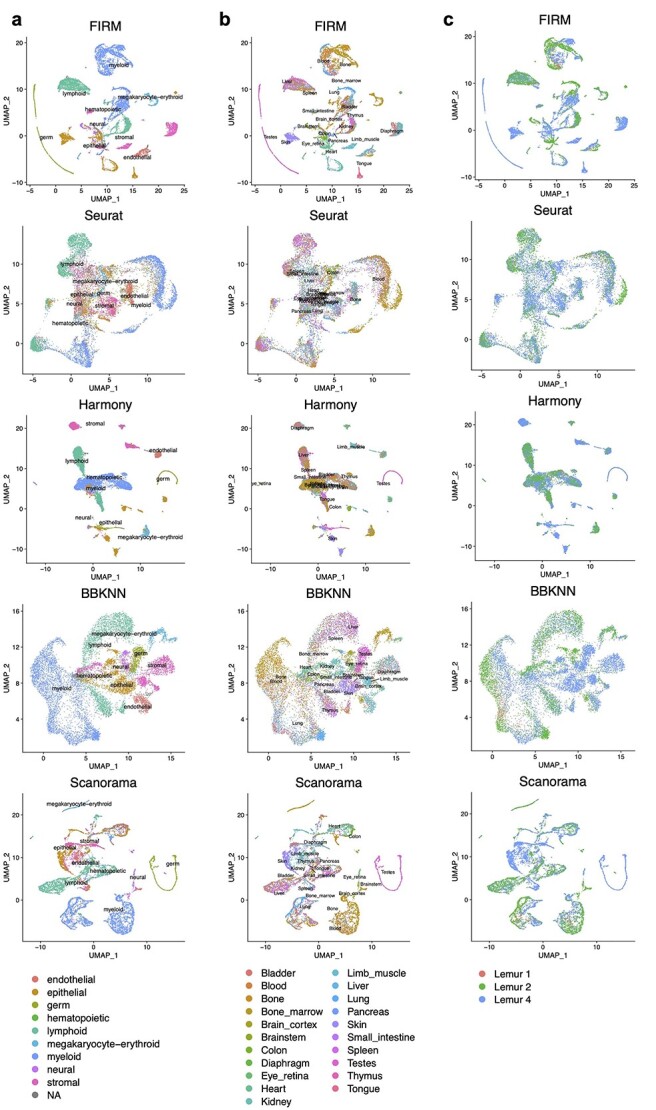
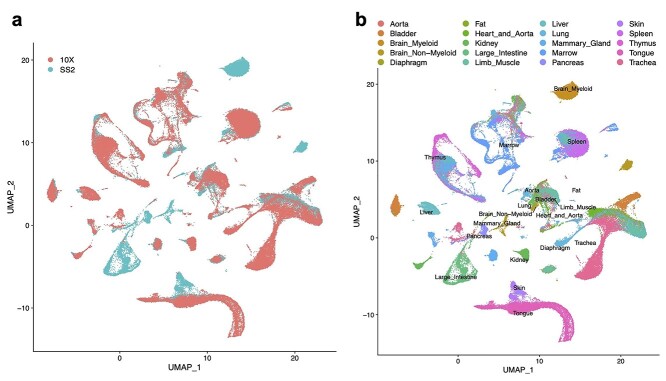
References
-
- Zilionis R, Nainys J, Veres A, et al. . Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc 2017;12(1):44–73. - PubMed
