

Nutrition interventions in congenital disorders of glycosylation
- PMID: 35562242
- PMCID: PMC9375550
- DOI: 10.1016/j.molmed.2022.04.003
Nutrition interventions in congenital disorders of glycosylation
Abstract
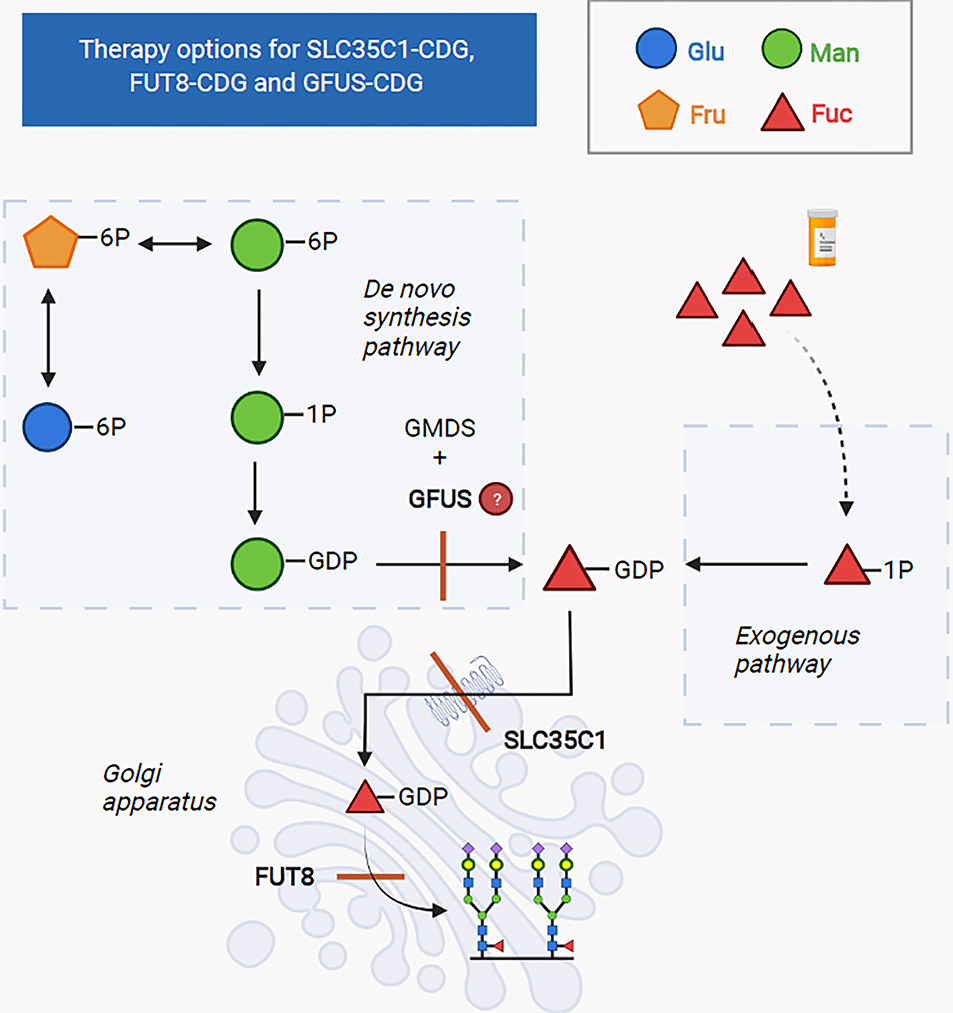
Congenital disorders of glycosylation (CDG) are a group of more than 160 inborn errors of metabolism affecting multiple pathways of protein and lipid glycosylation. Patients present with a wide range of symptoms and therapies are only available for very few subtypes. Specific nutritional treatment options for certain CDG types include oral supplementation of monosaccharide sugars, manganese, uridine, or pyridoxine. Additional management includes specific diets (i.e., complex carbohydrate or ketogenic diet), iron supplementation, and albumin infusions. We review the dietary management in CDG with a focus on two subgroups: N-linked glycosylation defects and GPI-anchor disorders.
Keywords: GPI-anchor disorder; N-linked CDG; hypoglycemia; manganese; monosaccharide therapy; pyridoxine.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests No interests are declared.
Figures


References
-
- Schjoldager KT et al. (2020) Global view of human protein glycosylation pathways and functions. Nature Reviews Molecular Cell Biology 21 (12), 729–749 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
