

Immune and Inflammatory Networks in Myocardial Infarction: Current Research and Its Potential Implications for the Clinic
- PMID: 35563605
- PMCID: PMC9102812
- DOI: 10.3390/ijms23095214
Immune and Inflammatory Networks in Myocardial Infarction: Current Research and Its Potential Implications for the Clinic
Abstract
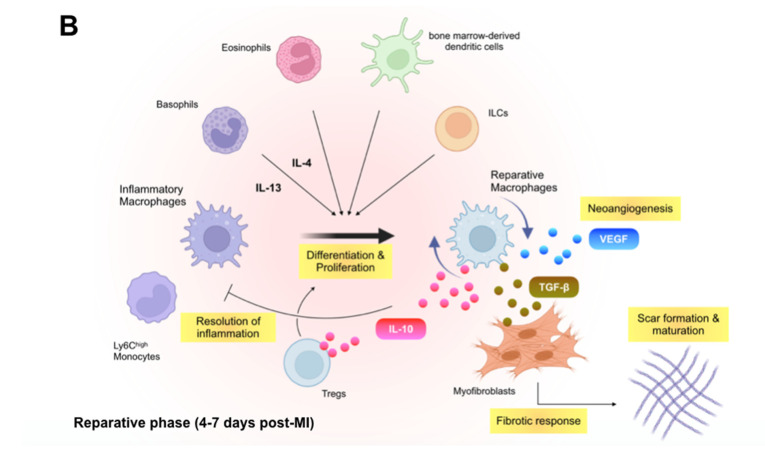
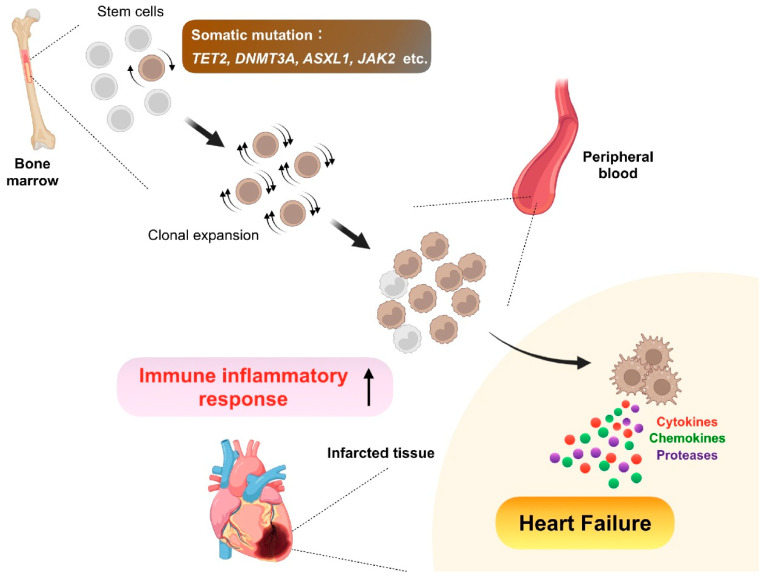
Despite recent scientific and technological advances, myocardial infarction (MI) still represents a major global health problem, leading to high morbidity and mortality worldwide. During the post-MI wound healing process, dysregulated immune inflammatory pathways and failure to resolve inflammation are associated with maladaptive left ventricular remodeling, progressive heart failure, and eventually poor outcomes. Given the roles of immune cells in the host response against tissue injury, understanding the involved cellular subsets, sources, and functions is essential for discovering novel therapeutic strategies that preserve the protective immune system and promote optimal healing. This review discusses the cellular effectors and molecular signals across multi-organ systems, which regulate the inflammatory and reparative responses after MI. Additionally, we summarize the recent clinical and preclinical data that propel conceptual revolutions in cardiovascular immunotherapy.
Keywords: adaptive immunity; chemokines; clinical trial; clonal hematopoiesis; cytokines; growth factors; heart failure; hematopoiesis; immune cells; immunotherapy; inflammation; innate immunity; myocardial infarction.
Conflict of interest statement
The authors have no conflict of interest to declare.
Figures


References
-
- Tsao C.W., Aday A.W., Almarzooq Z.I., Alonso A., Beaton A.Z., Bittencourt M.S., Boehme A.K., Buxton A.E., Carson A.P., Commodore-Mensah Y., et al. Heart Disease and Stroke Statistics-2022 Update: A Report from the American Heart Association. Circulation. 2022;145:e153–e639. - PubMed
-
- Frazier C.G., Alexander K.P., Newby L.K., Anderson S., Iverson E., Packer M., Cohn J., Goldstein S., Douglas P.S. Associations of gender and etiology with outcomes in heart failure with systolic dysfunction: A pooled analysis of 5 randomized control trials. J. Am. Coll. Cardiol. 2007;49:1450–1458. doi: 10.1016/j.jacc.2006.11.041. - DOI - PubMed
-
- Shore S., Grau-Sepulveda M.V., Bhatt D.L., Heidenreich P.A., Eapen Z.J., Hernandez A.F., Yancy C.W., Fonarow G.C. Characteristics, Treatments, and Outcomes of Hospitalized Heart Failure Patients Stratified by Etiologies of Cardiomyopathy. JACC Heart Fail. 2015;3:906–916. doi: 10.1016/j.jchf.2015.06.012. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
