

Computational NEXAFS Characterization of Molecular Model Systems for 2D Boroxine Frameworks
- PMID: 35564319
- PMCID: PMC9100003
- DOI: 10.3390/nano12091610
Computational NEXAFS Characterization of Molecular Model Systems for 2D Boroxine Frameworks
Abstract
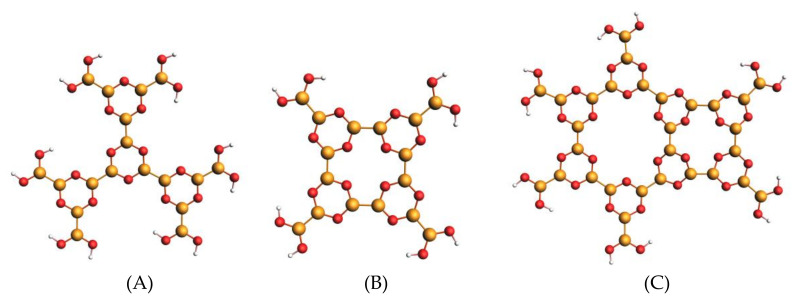
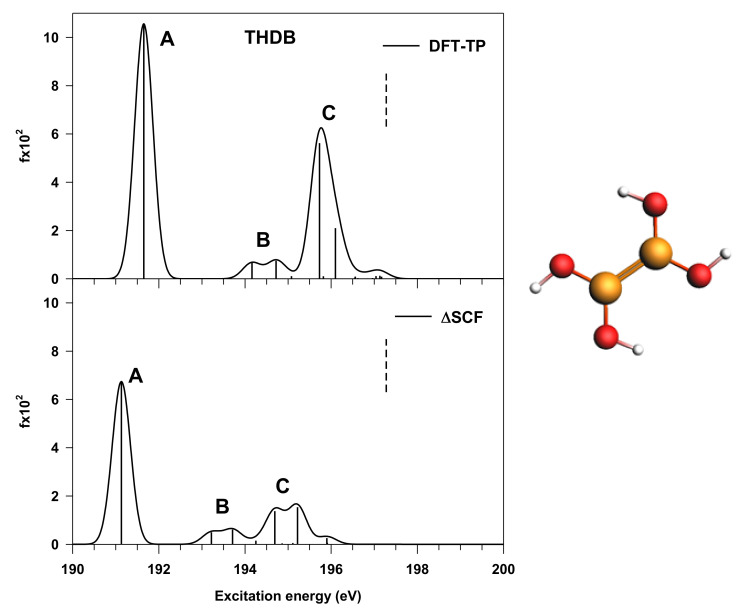
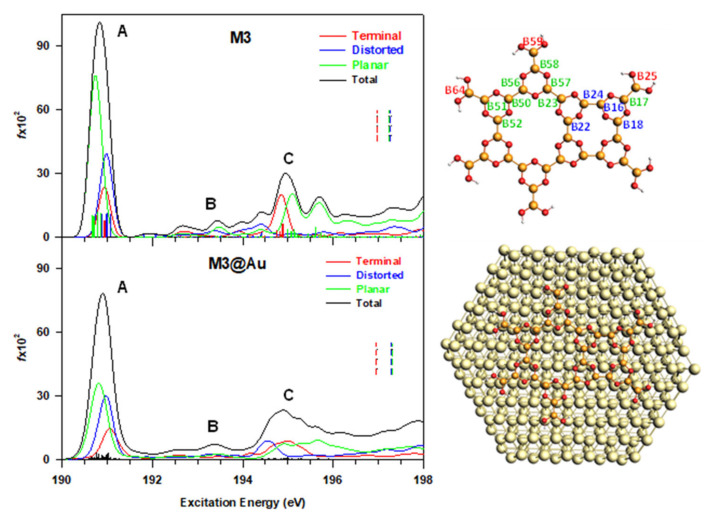
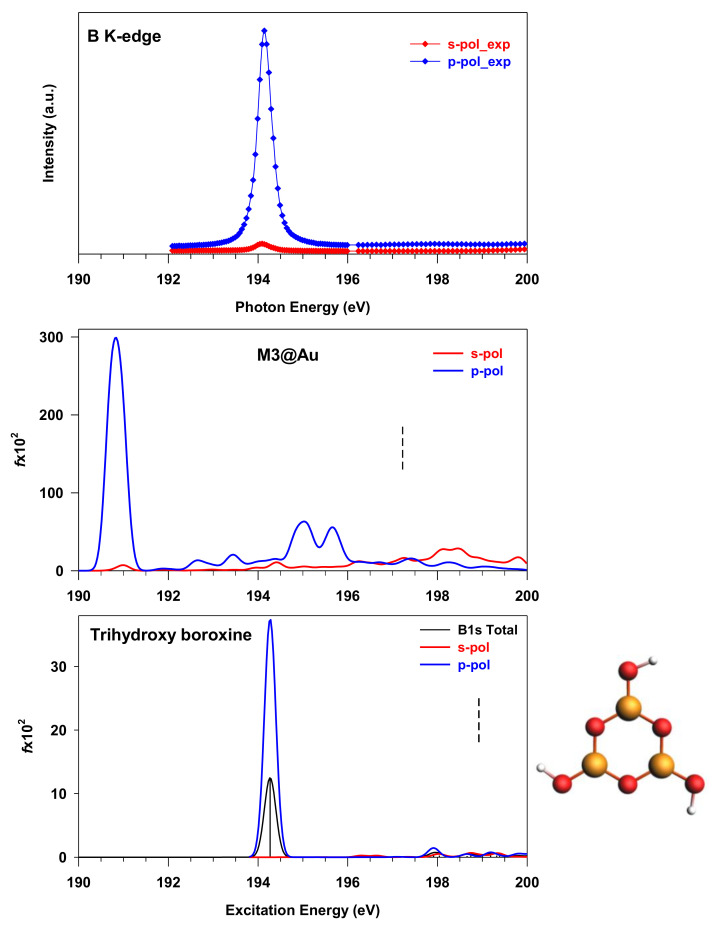
The electronic properties of 2D boroxine networks are computationally investigated by simulating the NEXAFS spectra of a series of molecular models, with or without morphologic defects, with respect to the ideal honeycomb structure. The models represent portions of an irregular 2D boroxine framework obtained experimentally, as supported by the Au(111) surface. The B K-edge NEXAFS spectra are calculated within the transition potential (TP) approximation (DFT-TP). The role of the Au(111) supporting surface on the spectral features has also been investigated by comparing the calculated spectra of a defect-rich model in its free-standing and supported form. The calculated NEXAFS spectra differ from the experimental ones, as the position of the main resonance does not match in the two cases. This finding could suggest the presence of a strong interaction of the 2D boroxine network with the Au substrate, which is not captured in the model calculations. However, good agreement between measured and calculated B K-edge NEXAFS spectra is obtained for a model system, namely, trihydroxy boroxine, in which the B atoms are less screened by the valence electrons compared to the B-B linked boroxine network models considered here. These results suggest catalytic activity in the gold substrate in promoting a weakening or even the breaking of the B-B bond, which is not revealed by calculations.
Keywords: DFT calculations; X-ray absorption spectroscopy; boroxine network.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Revealing the electronic properties of the B-B bond: the bis-catecholato diboron molecule.Phys Chem Chem Phys. 2021 Oct 27;23(41):23517-23525. doi: 10.1039/d1cp03428f. Phys Chem Chem Phys. 2021. PMID: 34642728
-
Carbon and Nitrogen K-Edge NEXAFS Spectra of Indole, 2,3-Dihydro-7-azaindole, and 3-Formylindole.J Phys Chem A. 2021 May 20;125(19):4160-4172. doi: 10.1021/acs.jpca.1c02570. Epub 2021 May 7. J Phys Chem A. 2021. PMID: 33961434
-
An efficient first principles method for molecular pump-probe NEXAFS spectra: Application to thymine and azobenzene.J Chem Phys. 2018 Oct 14;149(14):144112. doi: 10.1063/1.5050488. J Chem Phys. 2018. PMID: 30316280
-
Probing Intermolecular H-Bonding Interactions in Cyanuric Acid Networks: Quenching of the N K-Edge Sigma Resonances.J Phys Chem A. 2022 Oct 6;126(39):6870-6881. doi: 10.1021/acs.jpca.2c04517. Epub 2022 Sep 28. J Phys Chem A. 2022. PMID: 36168982 Free PMC article.
-
A detailed assignment of NEXAFS resonances of imidazolium based ionic liquids.Phys Chem Chem Phys. 2016 Mar 28;18(12):8654-61. doi: 10.1039/c5cp07434g. Phys Chem Chem Phys. 2016. PMID: 26948544
Cited by
-
Accurate Vertical Excitation Energies of BODIPY/Aza-BODIPY Derivatives from Excited-State Mean-Field Calculations.J Phys Chem A. 2022 Oct 13;126(40):7137-7146. doi: 10.1021/acs.jpca.2c04473. Epub 2022 Sep 29. J Phys Chem A. 2022. PMID: 36173265 Free PMC article.
References
-
- Dienstmaier J.F., Gigler A.M., Goetz A.J., Knochel P., Bein T., Lyapin A., Reichlmaier S., Heckl M., Lackinger W.M. Synthesis of Well-Ordered COF Monolayers: Surface Growth of Nanocrystalline Precursors versus Direct On-Surface Polycondensation. ACS Nano. 2011;5:9737–9745. doi: 10.1021/nn2032616. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
