

Infants and Newborns with Atypical Teratoid Rhabdoid Tumors (ATRT) and Extracranial Malignant Rhabdoid Tumors (eMRT) in the EU-RHAB Registry: A Unique and Challenging Population
- PMID: 35565313
- PMCID: PMC9100752
- DOI: 10.3390/cancers14092185
Infants and Newborns with Atypical Teratoid Rhabdoid Tumors (ATRT) and Extracranial Malignant Rhabdoid Tumors (eMRT) in the EU-RHAB Registry: A Unique and Challenging Population
Abstract
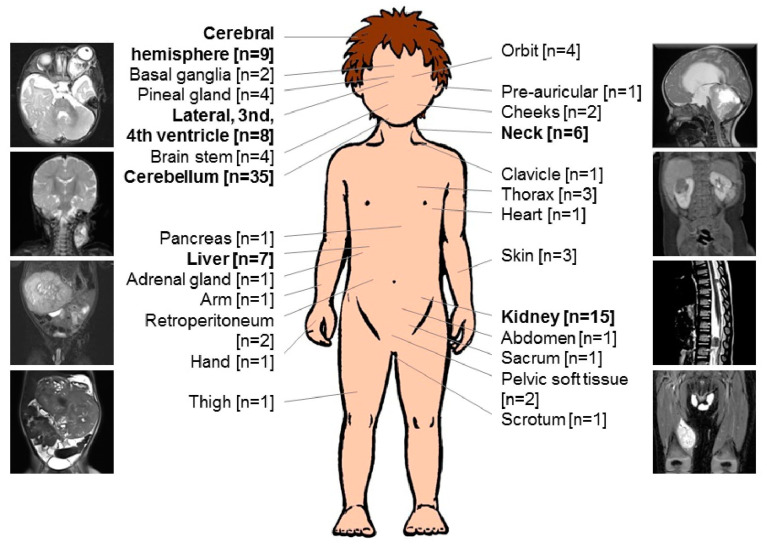
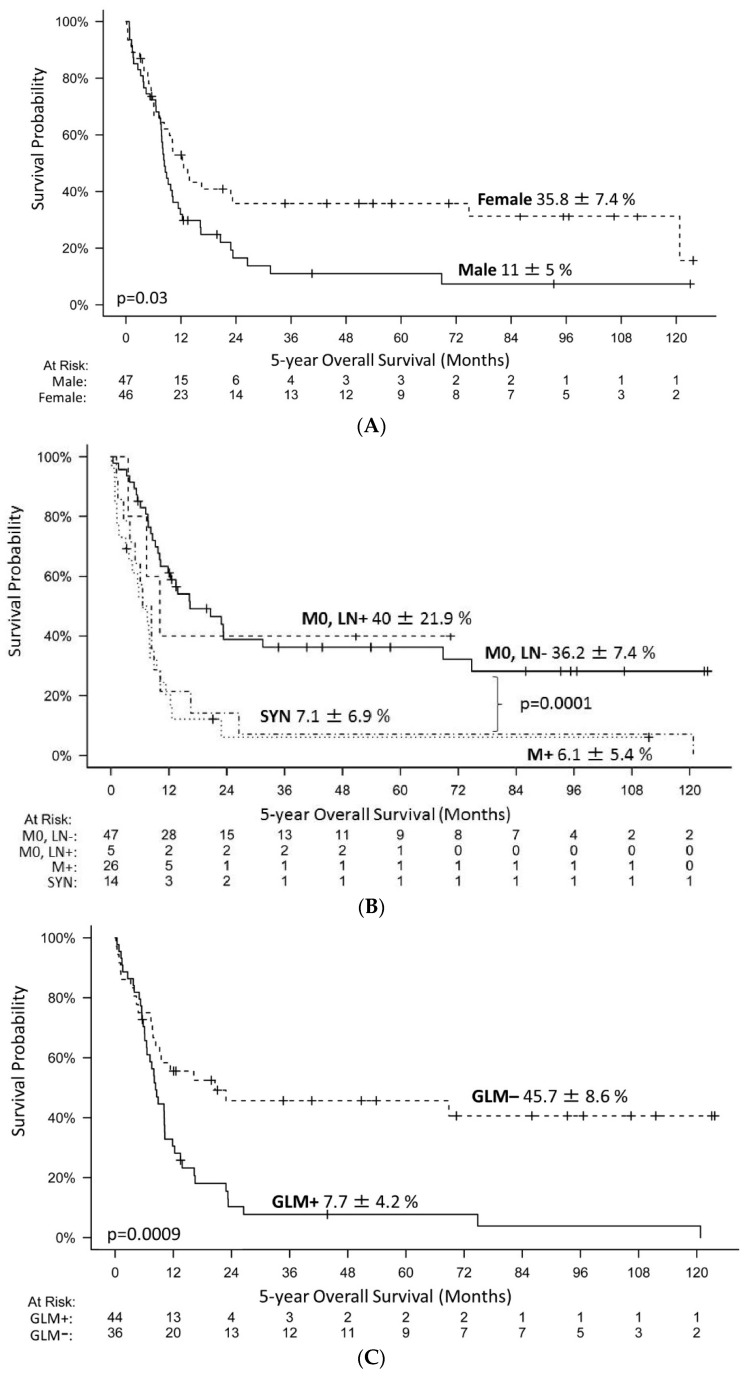
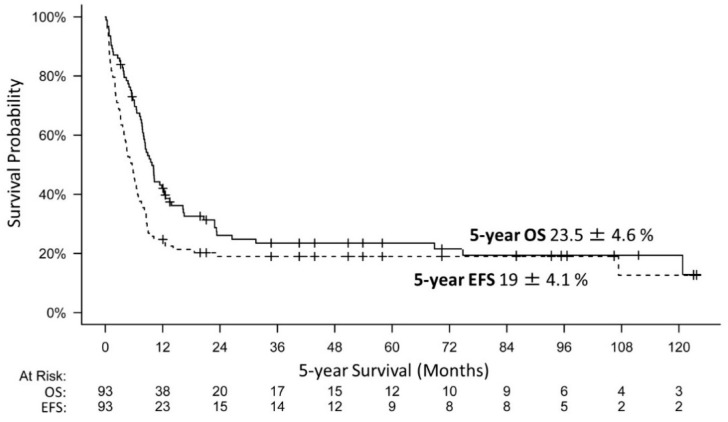
Introduction: Malignant rhabdoid tumors (MRT) predominantly affect infants and young children. Patients below six months of age represent a particularly therapeutically challenging group. Toxicity to developing organ sites limits intensity of treatment. Information on prognostic factors, genetics, toxicity of treatment and long-term outcomes is sparse. Methods: Clinical, genetic, and treatment data of 100 patients (aged below 6 months at diagnosis) from 13 European countries were analyzed (2005-2020). Tumors and matching blood samples were examined for SMARCB1 mutations using FISH, MLPA and Sanger sequencing. DNA methylation subgroups (ATRT-TYR, ATRT-SHH, and ATRT-MYC) were determined using 450 k / 850 k-profiling. Results: A total of 45 patients presented with ATRT, 29 with extracranial, extrarenal (eMRT) and 9 with renal rhabdoid tumors (RTK). Seventeen patients demonstrated synchronous tumors (SYN). Metastases (M+) were present in 27% (26/97) at diagnosis. A germline mutation (GLM) was detected in 55% (47/86). DNA methylation subgrouping was available in 50% (31 / 62) with ATRT or SYN; for eMRT, methylation-based subgrouping was not performed. The 5-year overall (OS) and event free survival (EFS) rates were 23.5 ± 4.6% and 19 ± 4.1%, respectively. Male sex (11 ± 5% vs. 35.8 ± 7.4%), M+ stage (6.1 ± 5.4% vs. 36.2 ± 7.4%), presence of SYN (7.1 ± 6.9% vs. 26.6 ± 5.3%) and GLM (7.7 ± 4.2% vs. 45.7 ± 8.6%) were significant prognostic factors for 5-year OS. Molecular subgrouping and survival analyses confirm a previously described survival advantage for ATRT-TYR. In an adjusted multivariate model, clinical factors that favorably influence the prognosis were female sex, localized stage, absence of a GLM and maintenance therapy. Conclusions: In this cohort of homogenously treated infants with MRT, significant predictors of outcome were sex, M-stage, GLM and maintenance therapy. We confirm the need to stratify which patient groups benefit from multimodal treatment, and which need novel therapeutic strategies. Biomarker-driven tailored trials may be a key option.
Keywords: EU-RHAB registry; RTPS1; RTPS2; SMARCB1; atypical teratoid rhabdoid tumors; extracranial malignant rhabdoid tumor; germline mutation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Cancer Statistics Reports for the Germany. [(accessed on 1 December 2021)]. Available online: http://www.Kinderkrebsregister.De/dkkr/ergebnisse/jahresberichte/jahresb....
-
- Reddy A.T., Strother D.R., Judkins A.R., Burger P.C., Pollack I.F., Krailo M.D., Buxton A.B., Williams-Hughes C., Fouladi M., Mahajan A., et al. Efficacy of high-dose chemotherapy and three-dimensional conformal radiation for atypical teratoid/rhabdoid tumor: A report from the children’s oncology group trial acns0333. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020;38:1175–1185. doi: 10.1200/JCO.19.01776. - DOI - PMC - PubMed
-
- Frühwald M.C., Hasselblatt M., Nemes K., Bens S., Steinbügl M., Johann P.D., Kerl K., Hauser P., Quiroga E., Solano-Paez P., et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro-Oncology. 2020;22:1006–1017. doi: 10.1093/neuonc/noz244. - DOI - PMC - PubMed
-
- Nemes K., Bens S., Kachanov D., Teleshova M., Hauser P., Simon T., Tippelt S., Woessmann W., Beck O., Flotho C., et al. Clinical and genetic risk factors define two risk groups of extracranial malignant rhabdoid tumours (emrt/rtk) Eur. J. Cancer. 2021;142:112–122. doi: 10.1016/j.ejca.2020.10.004. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
