

Coordination Chemistry of Nucleotides and Antivirally Active Acyclic Nucleoside Phosphonates, including Mechanistic Considerations
- PMID: 35565975
- PMCID: PMC9103026
- DOI: 10.3390/molecules27092625
Coordination Chemistry of Nucleotides and Antivirally Active Acyclic Nucleoside Phosphonates, including Mechanistic Considerations
Abstract
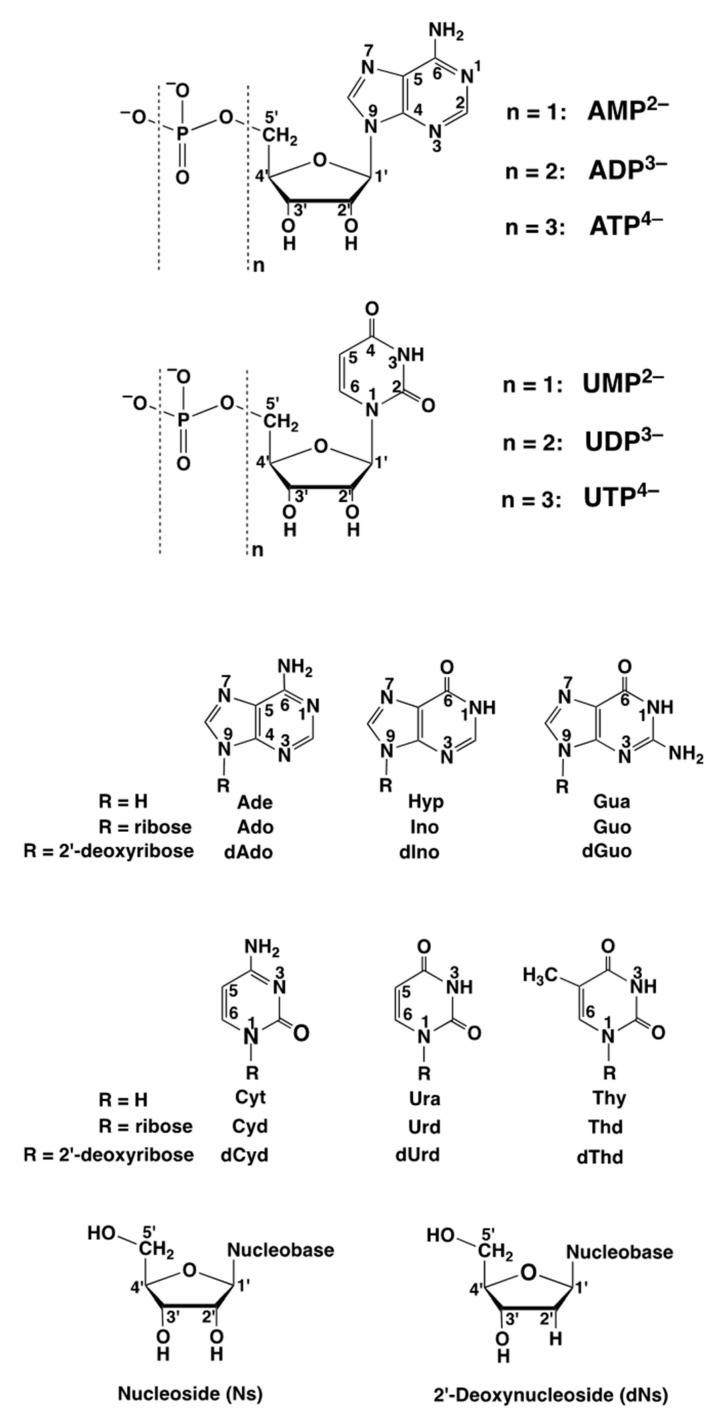
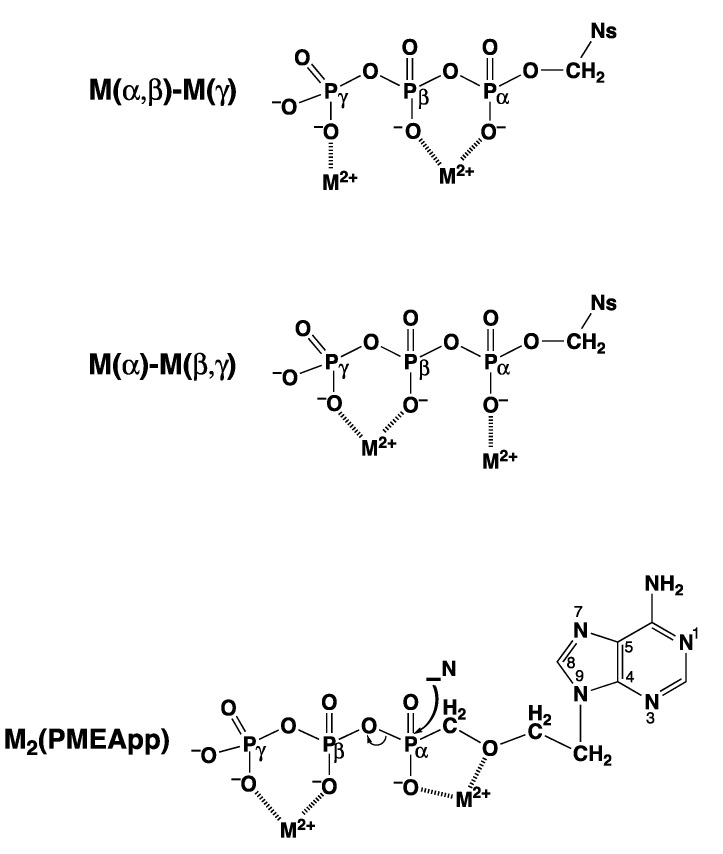
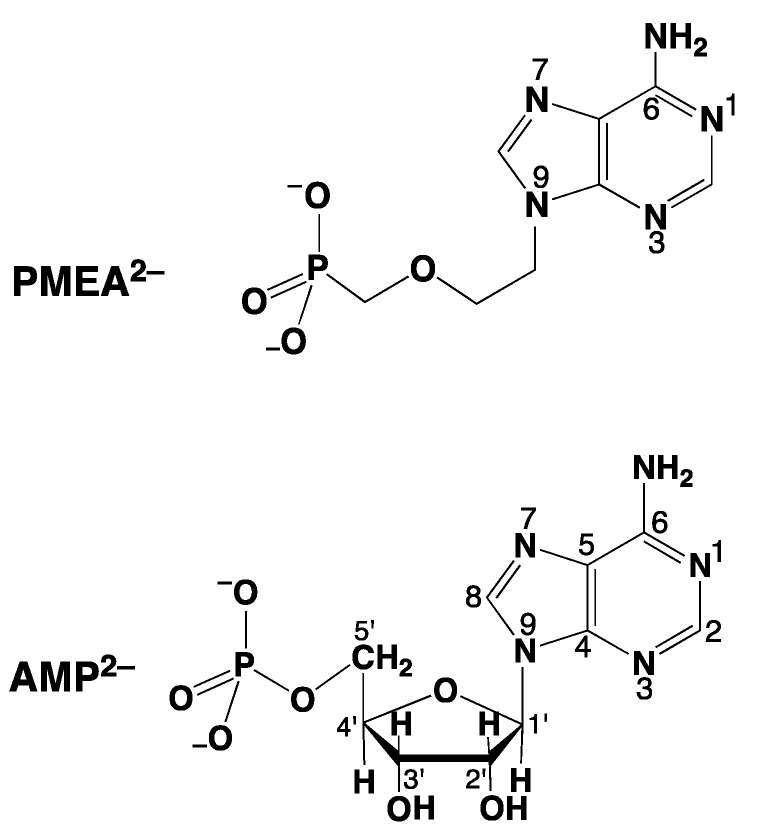
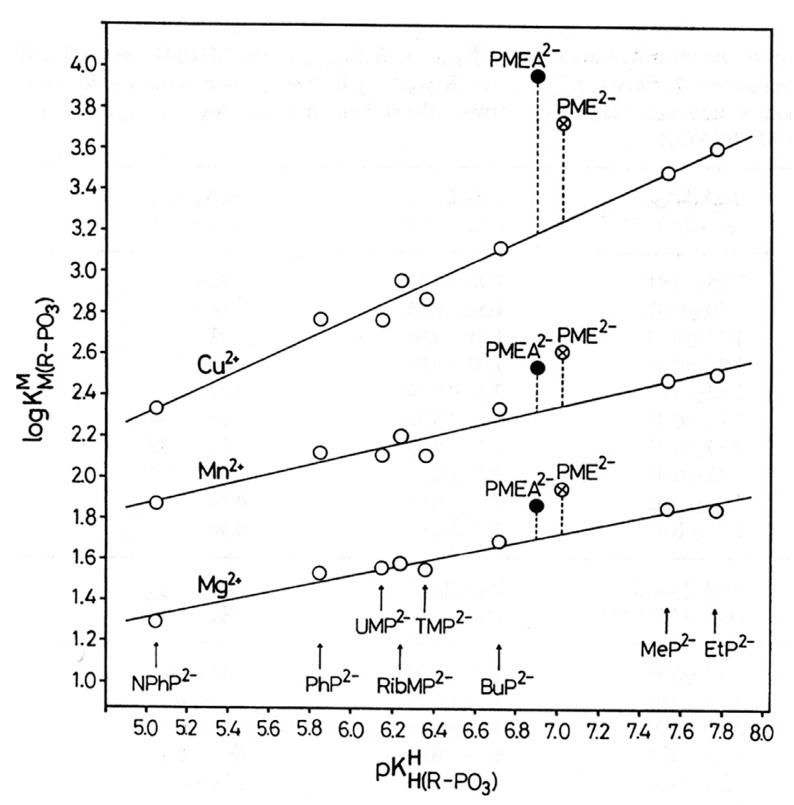
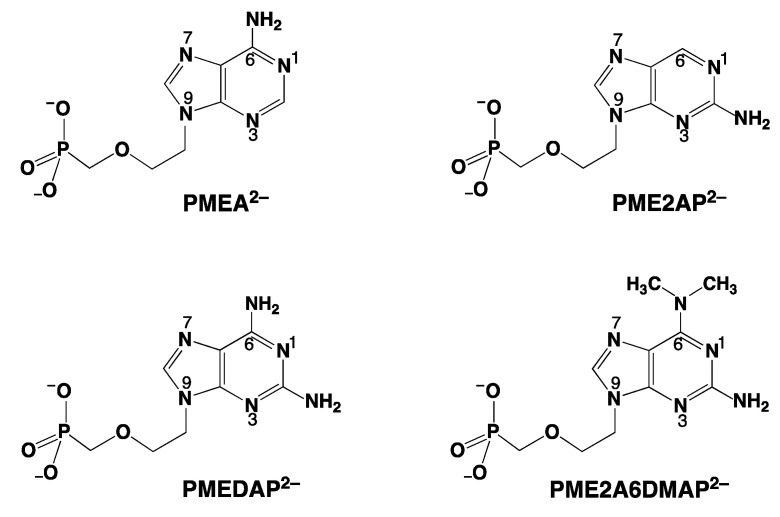
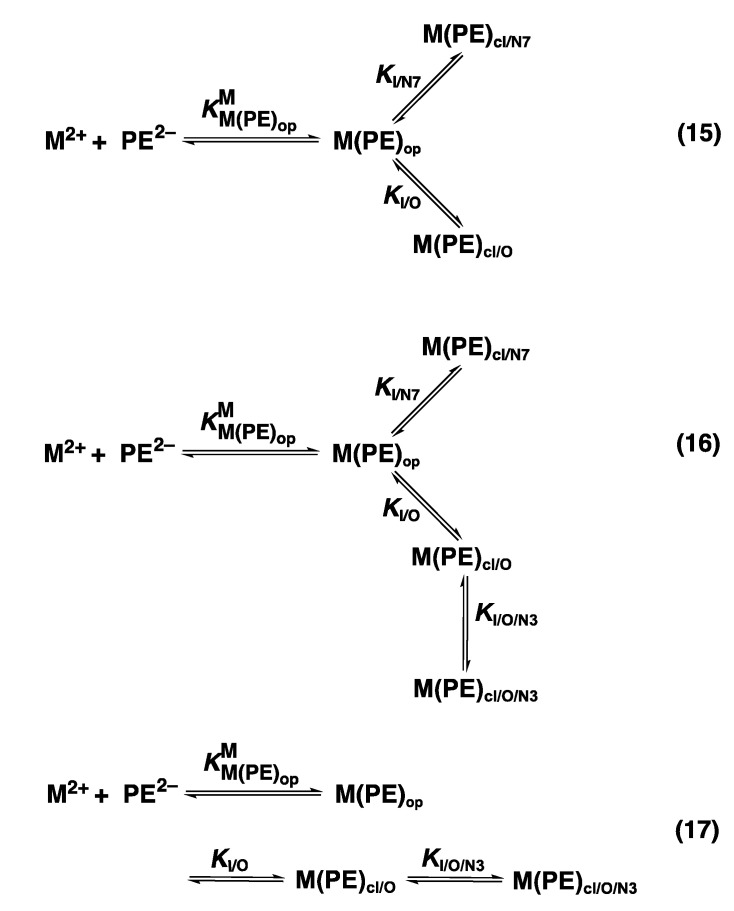
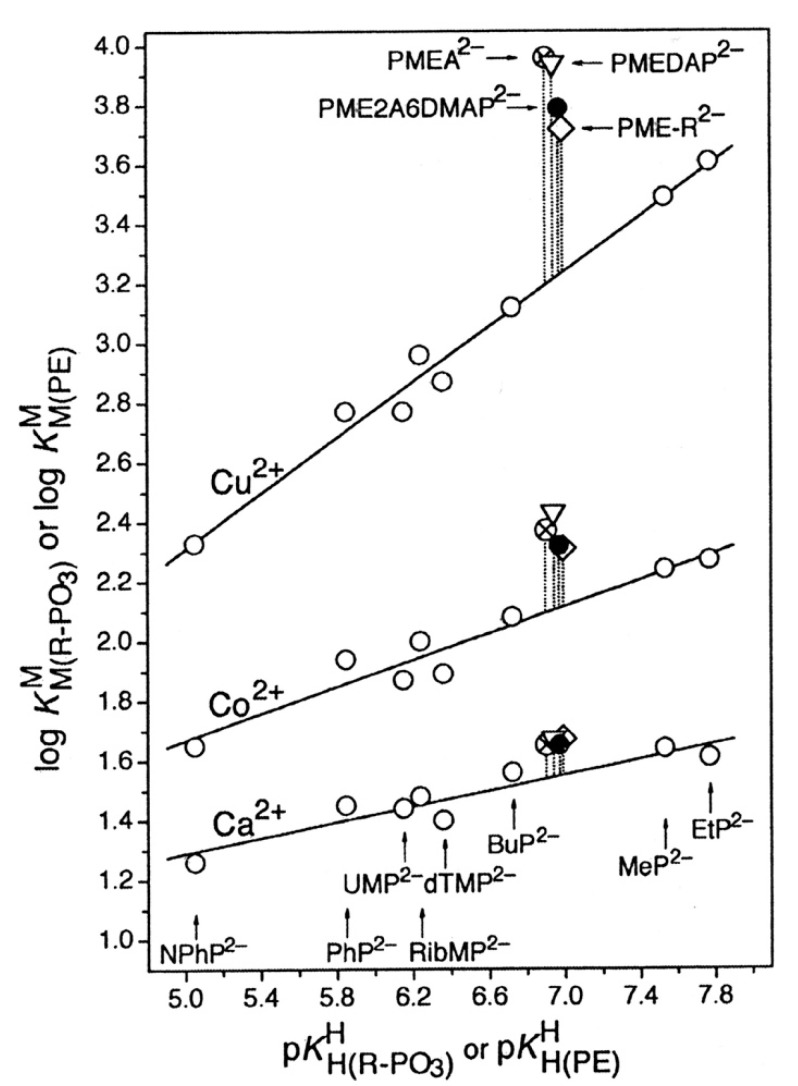
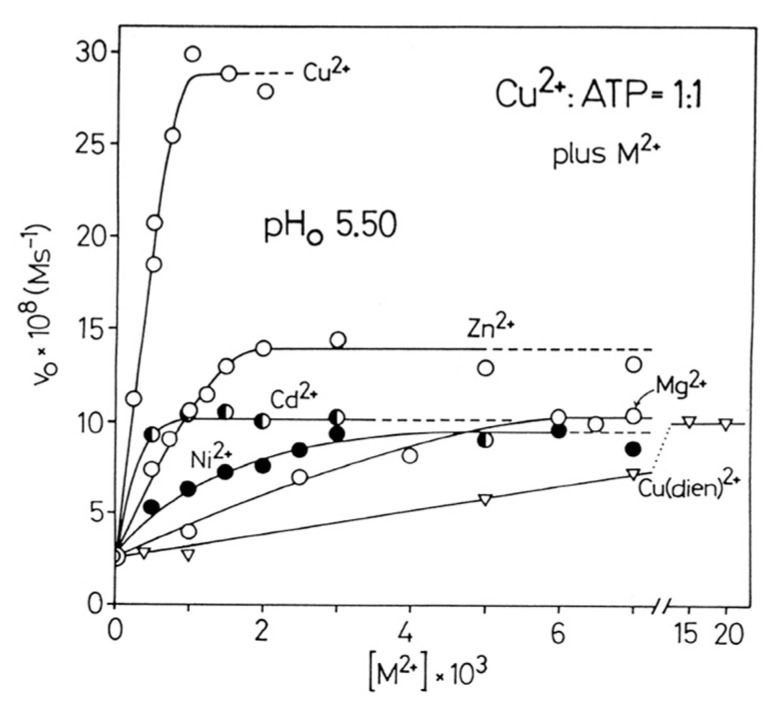
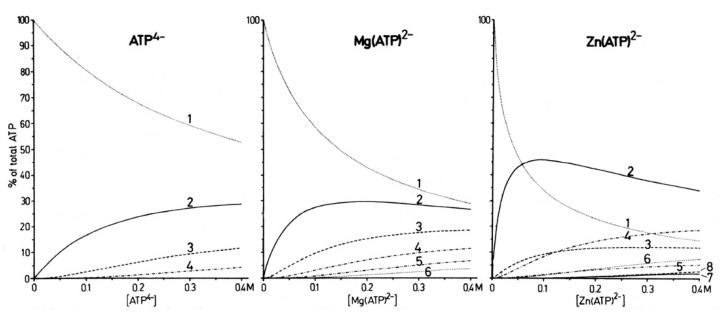
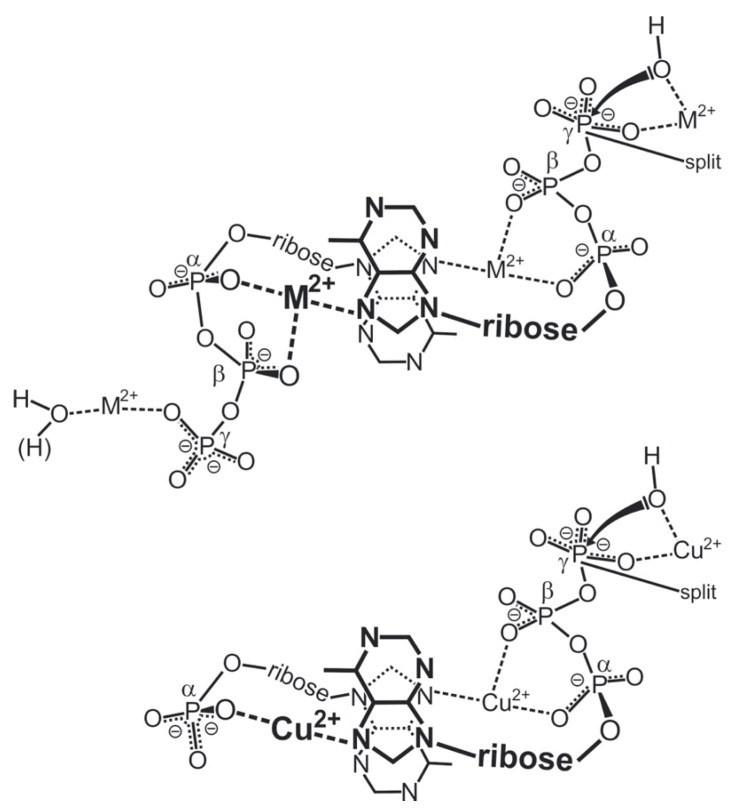
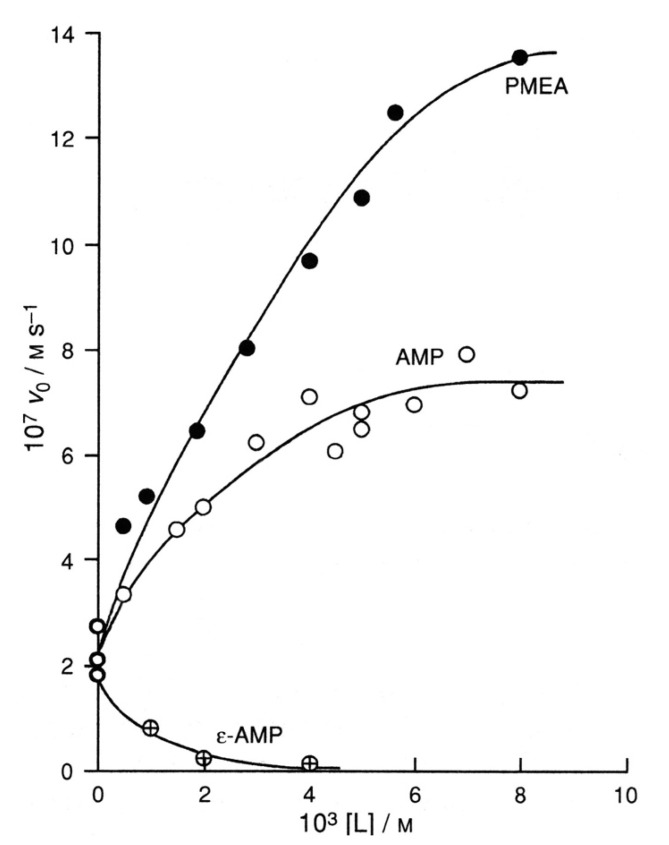
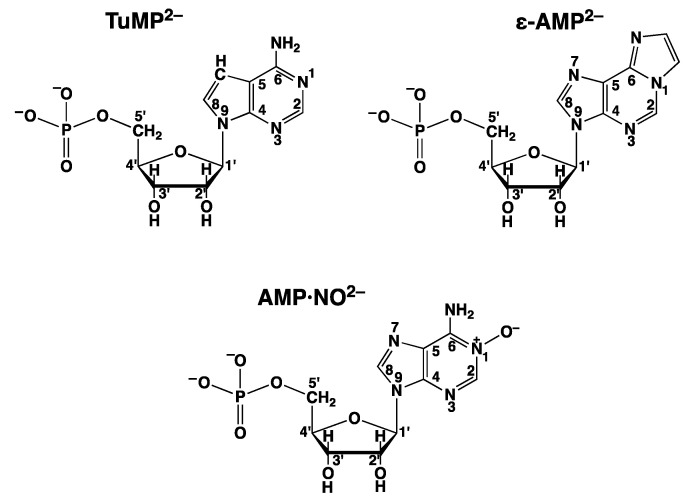
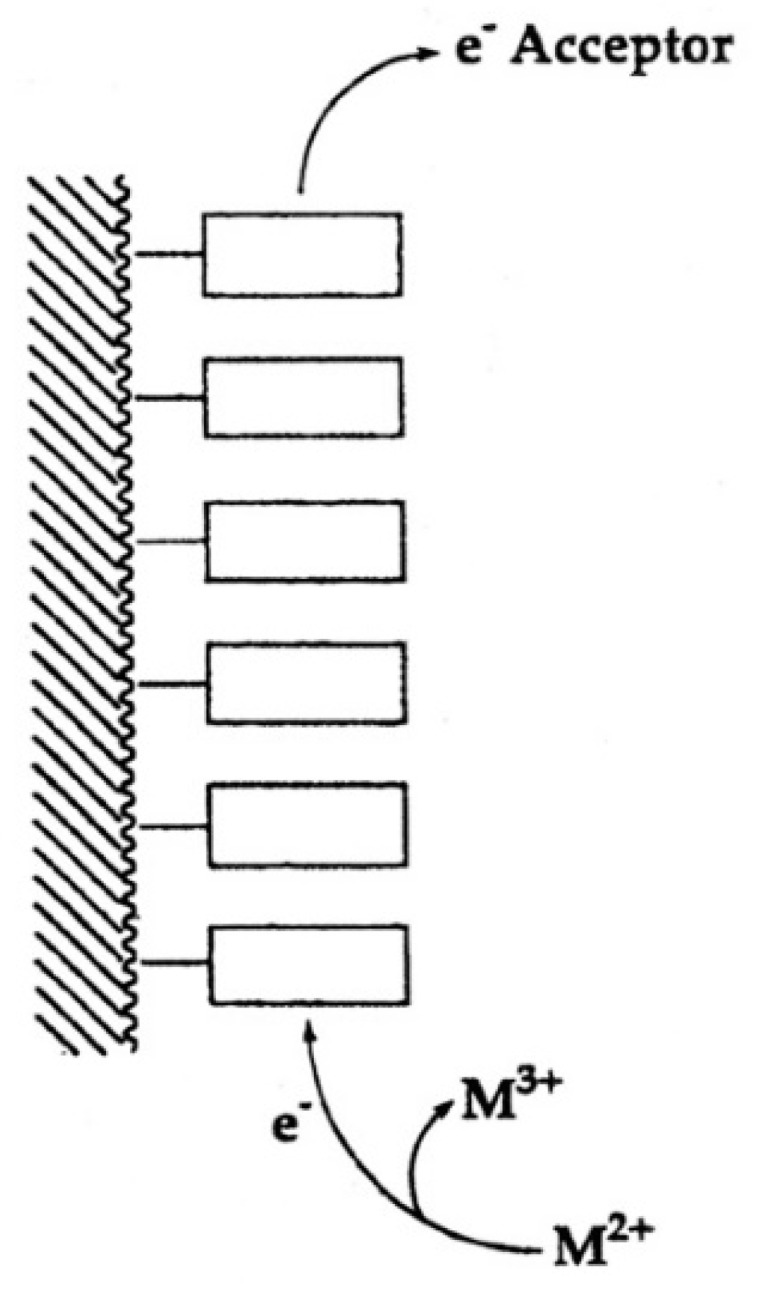
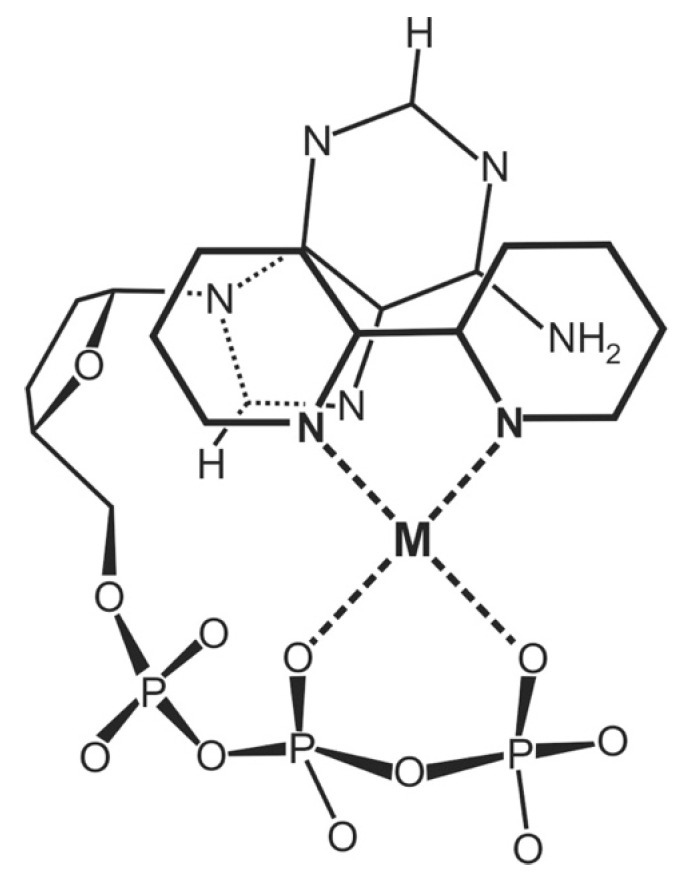
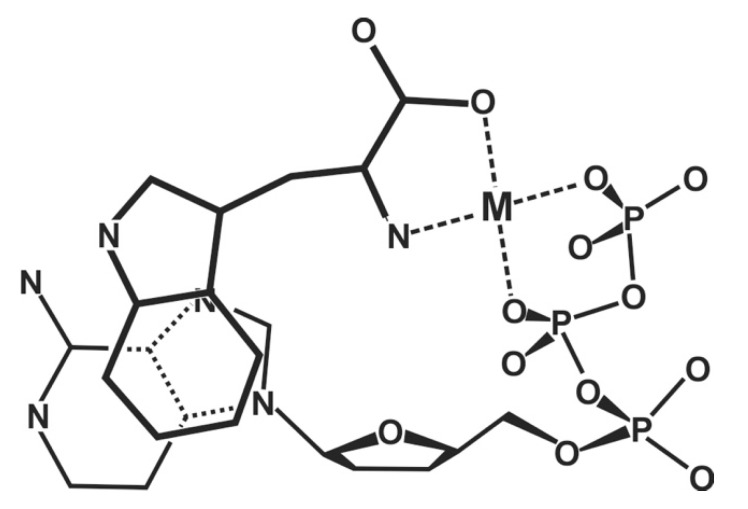
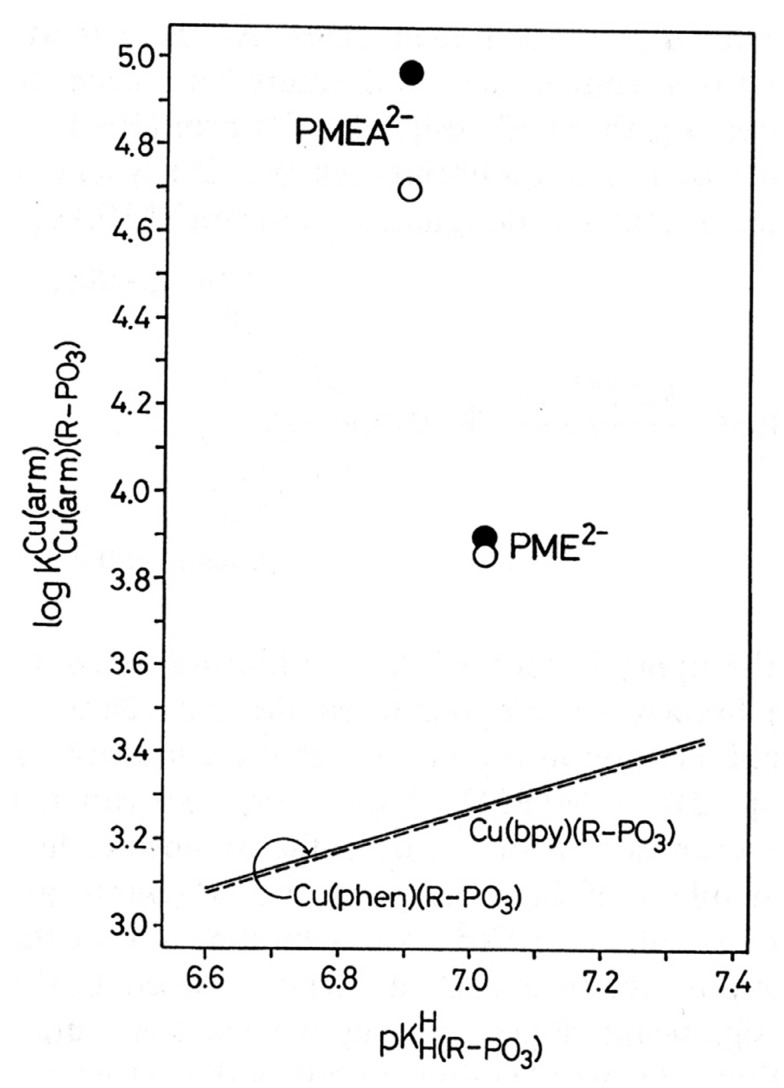
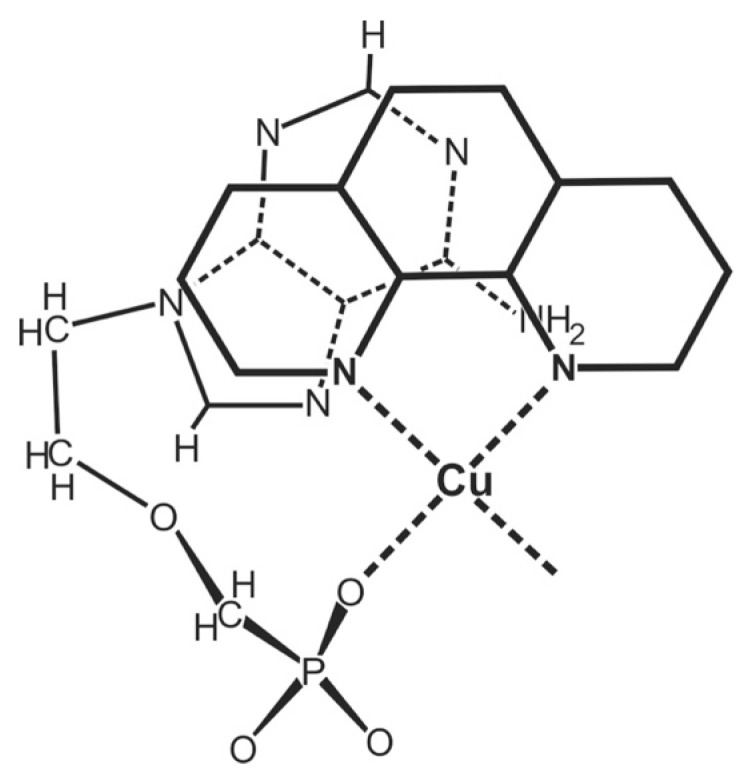
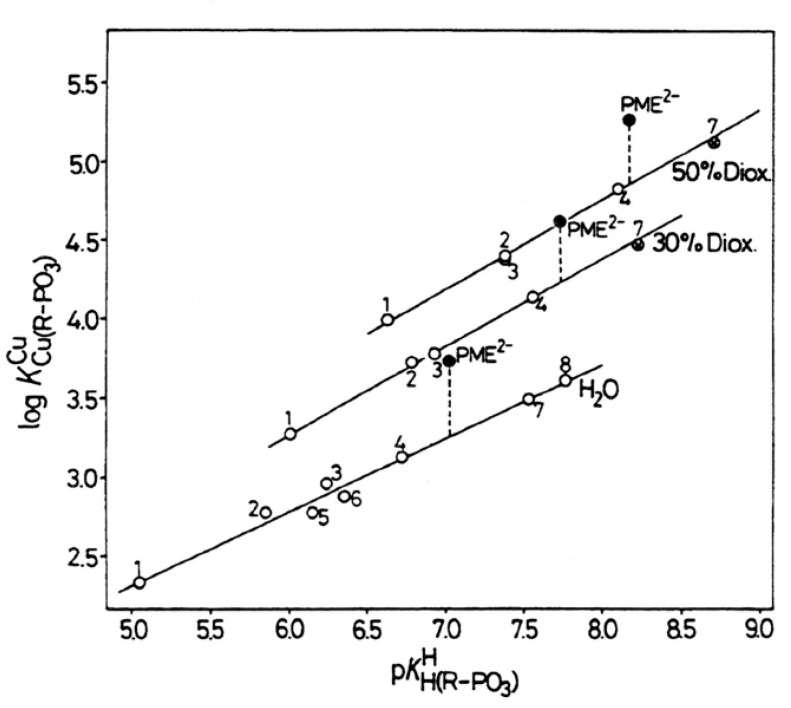
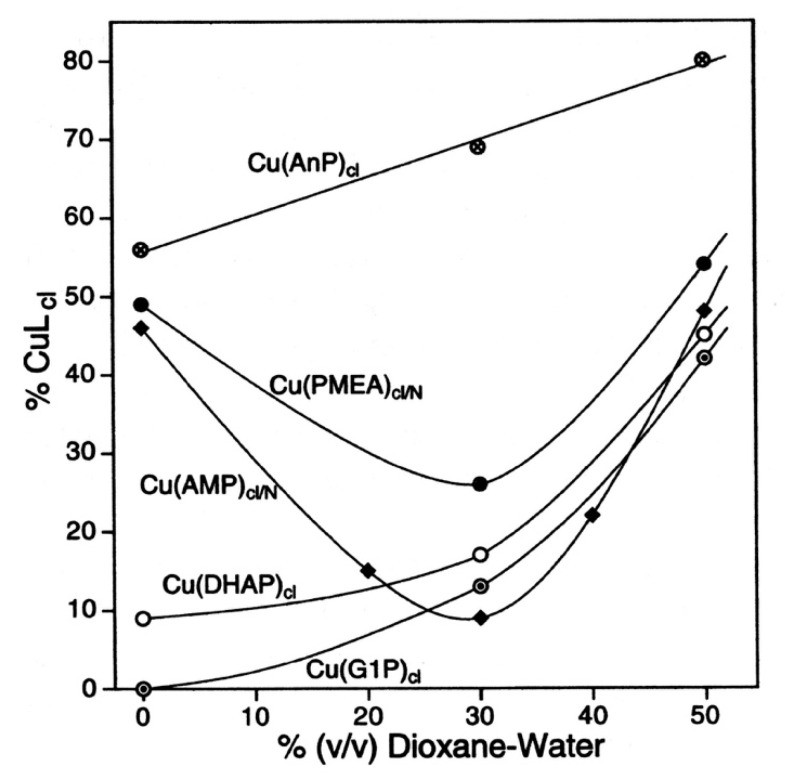
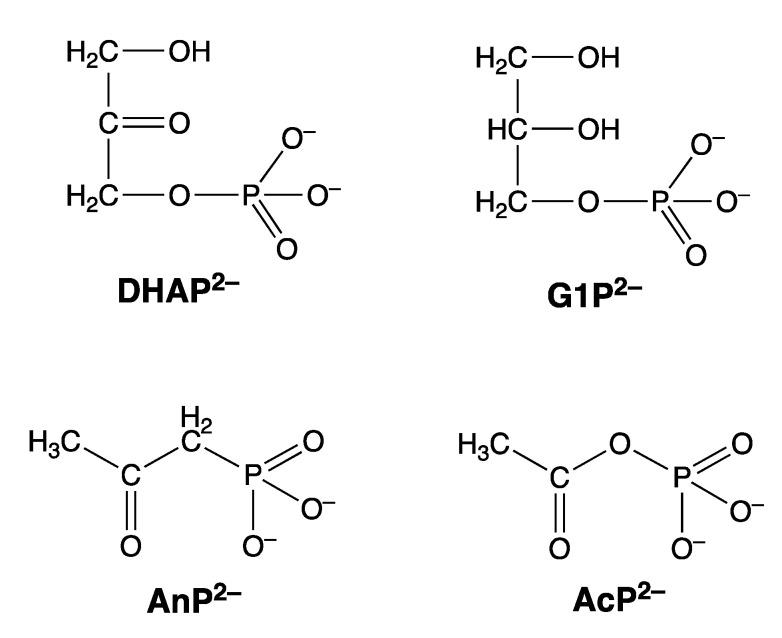
Considering that practically all reactions that involve nucleotides also involve metal ions, it is evident that the coordination chemistry of nucleotides and their derivatives is an essential corner stone of biological inorganic chemistry. Nucleotides are either directly or indirectly involved in all processes occurring in Nature. It is therefore no surprise that the constituents of nucleotides have been chemically altered-that is, at the nucleobase residue, the sugar moiety, and also at the phosphate group, often with the aim of discovering medically useful compounds. Among such derivatives are acyclic nucleoside phosphonates (ANPs), where the sugar moiety has been replaced by an aliphatic chain (often also containing an ether oxygen atom) and the phosphate group has been replaced by a phosphonate carrying a carbon-phosphorus bond to make the compounds less hydrolysis-sensitive. Several of these ANPs show antiviral activity, and some of them are nowadays used as drugs. The antiviral activity results from the incorporation of the ANPs into the growing nucleic acid chain-i.e., polymerases accept the ANPs as substrates, leading to chain termination because of the missing 3'-hydroxyl group. We have tried in this review to describe the coordination chemistry (mainly) of the adenine nucleotides AMP and ATP and whenever possible to compare it with that of the dianion of 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA2- = adenine(N9)-CH2-CH2-O-CH2-PO32) [or its diphosphate (PMEApp4-)] as a representative of the ANPs. Why is PMEApp4- a better substrate for polymerases than ATP4-? There are three reasons: (i) PMEA2- with its anti-like conformation (like AMP2-) fits well into the active site of the enzyme. (ii) The phosphonate group has an enhanced metal ion affinity because of its increased basicity. (iii) The ether oxygen forms a 5-membered chelate with the neighboring phosphonate and favors thus coordination at the Pα group. Research on ANPs containing a purine residue revealed that the kind and position of the substituent at C2 or C6 has a significant influence on the biological activity. For example, the shift of the (C6)NH2 group in PMEA to the C2 position leads to 9-[2-(phosphonomethoxy)ethyl]-2-aminopurine (PME2AP), an isomer with only a moderate antiviral activity. Removal of (C6)NH2 favors N7 coordination, e.g., of Cu2+, whereas the ether O atom binding of Cu2+ in PMEA facilitates N3 coordination via adjacent 5- and 7-membered chelates, giving rise to a Cu(PMEA)cl/O/N3 isomer. If the metal ions (M2+) are M(α,β)-M(γ)-coordinated at a triphosphate chain, transphosphorylation occurs (kinases, etc.), whereas metal ion binding in a M(α)-M(β,γ)-type fashion is relevant for polymerases. It may be noted that with diphosphorylated PMEA, (PMEApp4-), the M(α)-M(β,γ) binding is favored because of the formation of the 5-membered chelate involving the ether O atom (see above). The self-association tendency of purines leads to the formation of dimeric [M2(ATP)]2(OH)- stacks, which occur in low concentration and where one half of the molecule undergoes the dephosphorylation reaction and the other half stabilizes the structure-i.e., acts as the "enzyme" by bridging the two ATPs. In accord herewith, one may enhance the reaction rate by adding AMP2- to the [Cu2(ATP)]2(OH)- solution, as this leads to the formation of mixed stacked Cu3(ATP)(AMP)(OH)- species, in which AMP2- takes over the structuring role, while the other "half" of the molecule undergoes dephosphorylation. It may be added that Cu3(ATP)(PMEA) or better Cu3(ATP)(PMEA)(OH)- is even a more reactive species than Cu3(ATP)(AMP)(OH)-. - The matrix-assisted self-association and its significance for cell organelles with high ATP concentrations is summarized and discussed, as is, e.g., the effect of tryptophanate (Trp-), which leads to the formation of intramolecular stacks in M(ATP)(Trp)3- complexes (formation degree about 75%). Furthermore, it is well-known that in the active-site cavities of enzymes the dielectric constant, compared with bulk water, is reduced; therefore, we have summarized and discussed the effect of a change in solvent polarity on the stability and structure of binary and ternary complexes: Opposite effects on charged O sites and neutral N sites are observed, and this leads to interesting insights.
Keywords: 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA); acyclic nucleoside phosphonates; antivirals; cell organelles; competing solvent effects; complex stabilities; dephosphorylation; hydrolysis of ATP; intramolecular equilibria; isodesmic model; kinases; mechanistic considerations; metal ion complexes; mixed ligand complexes; nucleic acids; nucleotide analogues; polarity changes; polymerases; self-association; solvent effects; ternary complexes; triphosphate coordination modes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Extent of intramolecular π-stacks in aqueous solution in mixed-ligand copper(II) complexes formed by heteroaromatic amines and several 2-aminopurine derivatives of the antivirally active nucleotide analog 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA).Chem Biodivers. 2012 Sep;9(9):2008-34. doi: 10.1002/cbdv.201200022. Chem Biodivers. 2012. PMID: 22976988
-
Quantification of isomeric equilibria formed by metal ion complexes of 8-[2-(phosphonomethoxy)ethyl]-8-azaadenine (8,8aPMEA) and 9-[2-(phosphonomethoxy)ethyl]-8-azaadenine (9,8aPMEA). Derivatives of the antiviral nucleotide analogue 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA).J Biol Inorg Chem. 2004 Dec;9(8):961-72. doi: 10.1007/s00775-004-0591-7. Epub 2004 Oct 20. J Biol Inorg Chem. 2004. PMID: 15503234
-
Metal ion-binding properties of 9-[(2-phosphonomethoxy)ethyl]-2-aminopurine (PME2AP), an isomer of the antiviral nucleotide analogue 9-[(2-phosphonomethoxy)ethyl]adenine (PMEA). Steric guiding of metal ion-coordination by the purine-amino group.Dalton Trans. 2010 Jul 21;39(27):6344-54. doi: 10.1039/c005238h. Epub 2010 Jun 3. Dalton Trans. 2010. PMID: 20523923
-
Metal ion complexes of antivirally active nucleotide analogues. Conclusions regarding their biological action.Chem Soc Rev. 2004 Mar 30;33(3):191-200. doi: 10.1039/b310349h. Epub 2004 Feb 10. Chem Soc Rev. 2004. PMID: 15026824 Review.
-
Complex formation of cadmium with sugar residues, nucleobases, phosphates, nucleotides, and nucleic acids.Met Ions Life Sci. 2013;11:191-274. doi: 10.1007/978-94-007-5179-8_8. Met Ions Life Sci. 2013. PMID: 23430775 Review.
Cited by
-
Platinum-Nucleos(t)ide Compounds as Possible Antimetabolites for Antitumor/Antiviral Therapy: Properties and Perspectives.Pharmaceutics. 2023 Mar 14;15(3):941. doi: 10.3390/pharmaceutics15030941. Pharmaceutics. 2023. PMID: 36986802 Free PMC article. Review.
-
Propentofylline Improves Thiol-Based Antioxidant Defenses and Limits Lipid Peroxidation following Gliotoxic Injury in the Rat Brainstem.Biomedicines. 2023 Jun 7;11(6):1652. doi: 10.3390/biomedicines11061652. Biomedicines. 2023. PMID: 37371747 Free PMC article.
References
-
- Sigel A., Sigel H., editors. Metal Ions in Biological Systems. Volume 32. Marcel Dekker, Inc.; New York, NY, USA: Basel, Switzerland: Hong Kong, China: 1996. Interactions of Metal Ions with Nucleotides, Nucleid Acids, and Their Constituents; pp. 1–814.
-
- Sigel A., Sigel H., Sigel R.K.O., editors. Metal Ions in Life Sciences. Volume 9. Royal Society of Chemistry; Cambridge, UK: 2011. Structural and Catalytic Roles of Metal Ions in RNA; pp. 1–391. - PubMed
-
- Sigel A., Sigel H., Sigel R.K.O., editors. Metal Ions in Life Sciences. Volume 10. Springer; Dordrecht, The Netherlands: 2012. Interplay Between Metal Ions and Nucleic Acids; pp. 1–351.
-
- Aoki K. General Conclusions from Solid State Studies of Nucleotide-Metal Ion Complexes. Met. Ions Biol. Syst. 1996;32:91–134.
-
- Martin R.B., Mariam Y.H. Interactions between Metal Ions and Nucleic Bases, Nucleosides, and Nucleotides in Solution. Met. Ions Biol. Syst. 1979;8:57–124.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
