

Accelerating AutoDock Vina with GPUs
- PMID: 35566391
- PMCID: PMC9103882
- DOI: 10.3390/molecules27093041
Accelerating AutoDock Vina with GPUs
Abstract
AutoDock Vina is one of the most popular molecular docking tools. In the latest benchmark CASF-2016 for comparative assessment of scoring functions, AutoDock Vina won the best docking power among all the docking tools. Modern drug discovery is facing a common scenario of large virtual screening of drug hits from huge compound databases. Due to the seriality characteristic of the AutoDock Vina algorithm, there is no successful report on its parallel acceleration with GPUs. Current acceleration of AutoDock Vina typically relies on the stack of computing power as well as the allocation of resource and tasks, such as the VirtualFlow platform. The vast resource expenditure and the high access threshold of users will greatly limit the popularity of AutoDock Vina and the flexibility of its usage in modern drug discovery. In this work, we proposed a new method, Vina-GPU, for accelerating AutoDock Vina with GPUs, which is greatly needed for reducing the investment for large virtual screens and also for wider application in large-scale virtual screening on personal computers, station servers or cloud computing, etc. Our proposed method is based on a modified Monte Carlo using simulating annealing AI algorithm. It greatly raises the number of initial random conformations and reduces the search depth of each thread. Moreover, a classic optimizer named BFGS is adopted to optimize the ligand conformations during the docking progress, before a heterogeneous OpenCL implementation was developed to realize its parallel acceleration leveraging thousands of GPU cores. Large benchmark tests show that Vina-GPU reaches an average of 21-fold and a maximum of 50-fold docking acceleration against the original AutoDock Vina while ensuring their comparable docking accuracy, indicating its potential for pushing the popularization of AutoDock Vina in large virtual screens.
Keywords: AutoDock Vina; GPU; OpenCL; Vina-GPU.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
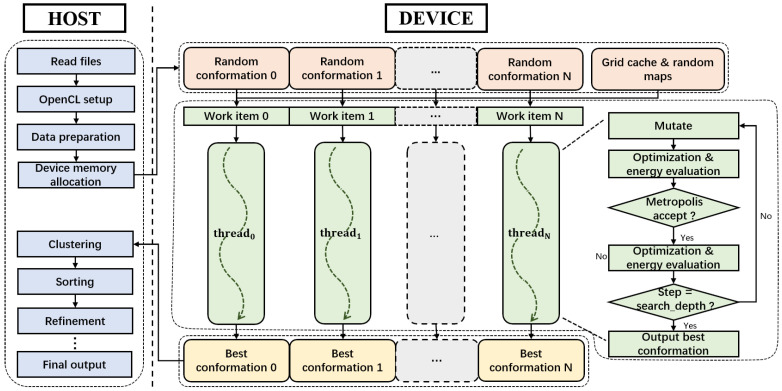
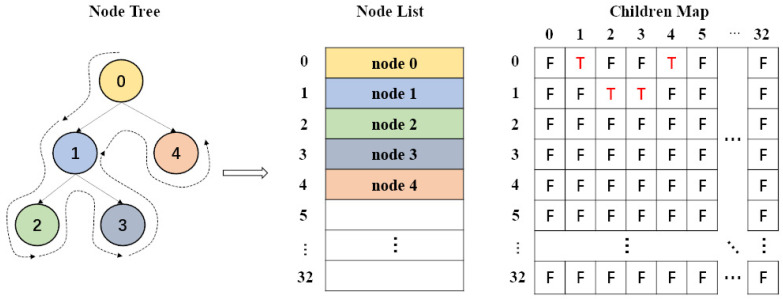


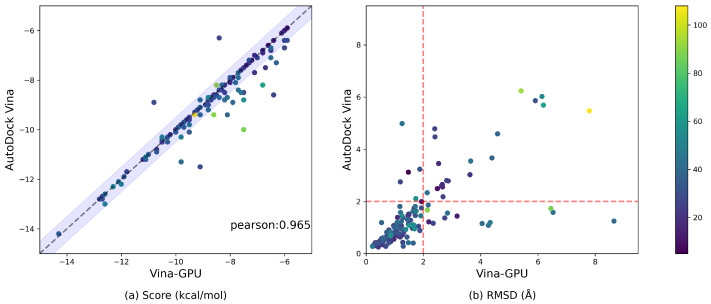
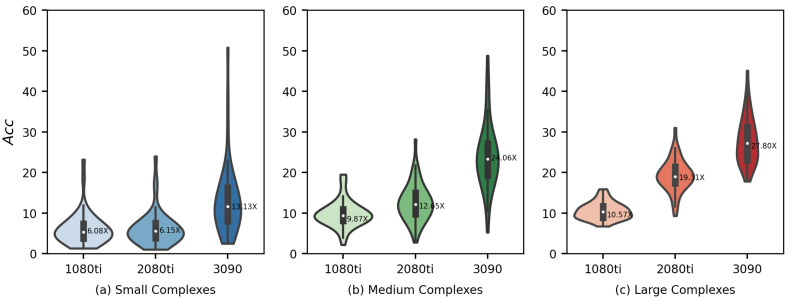







Similar articles
-
Vina-GPU 2.0: Further Accelerating AutoDock Vina and Its Derivatives with Graphics Processing Units.J Chem Inf Model. 2023 Apr 10;63(7):1982-1998. doi: 10.1021/acs.jcim.2c01504. Epub 2023 Mar 20. J Chem Inf Model. 2023. PMID: 36941232
-
Vina-GPU 2.1: Towards Further Optimizing Docking Speed and Precision of AutoDock Vina and Its Derivatives.IEEE/ACM Trans Comput Biol Bioinform. 2024 Nov-Dec;21(6):2382-2393. doi: 10.1109/TCBB.2024.3467127. Epub 2024 Dec 10. IEEE/ACM Trans Comput Biol Bioinform. 2024. PMID: 39320991
-
Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark.J Chem Inf Model. 2018 Aug 27;58(8):1697-1706. doi: 10.1021/acs.jcim.8b00312. Epub 2018 Jul 25. J Chem Inf Model. 2018. PMID: 29989806
-
Pose Classification Using Three-Dimensional Atomic Structure-Based Neural Networks Applied to Ion Channel-Ligand Docking.J Chem Inf Model. 2022 May 23;62(10):2301-2315. doi: 10.1021/acs.jcim.1c01510. Epub 2022 Apr 21. J Chem Inf Model. 2022. PMID: 35447030 Free PMC article. Review.
-
Survey of public domain software for docking simulations and virtual screening.Hum Genomics. 2011 Jul;5(5):497-505. doi: 10.1186/1479-7364-5-5-497. Hum Genomics. 2011. PMID: 21807604 Free PMC article. Review.
Cited by
-
Reprogramming the Transition States to Enhance C-N Cleavage Efficiency of Rhodococcus opacusl-Amino Acid Oxidase.JACS Au. 2024 Jan 16;4(2):557-569. doi: 10.1021/jacsau.3c00672. eCollection 2024 Feb 26. JACS Au. 2024. PMID: 38425913 Free PMC article.
-
Novel Flurbiprofen Derivatives as Antioxidant and Anti-Inflammatory Agents: Synthesis, In Silico, and In Vitro Biological Evaluation.Molecules. 2024 Jan 12;29(2):385. doi: 10.3390/molecules29020385. Molecules. 2024. PMID: 38257299 Free PMC article.
-
Computational drug repurposing for tuberculosis by inhibiting Ag85 complex proteins.Narra J. 2025 Apr;5(1):e1130. doi: 10.52225/narra.v5i1.1130. Epub 2025 Jan 17. Narra J. 2025. PMID: 40352212 Free PMC article.
-
Stereoselective Biosynthesis of Bichiral Amino Alcohols from Diketone via Difunctionalization.ACS Omega. 2025 May 27;10(22):23409-23420. doi: 10.1021/acsomega.5c01864. eCollection 2025 Jun 10. ACS Omega. 2025. PMID: 40521437 Free PMC article.
-
Rationally Designed Novel Antimicrobial Peptides Targeting Chitin Synthase for Combating Soybean Phytophthora Blight.Int J Mol Sci. 2024 Mar 20;25(6):3512. doi: 10.3390/ijms25063512. Int J Mol Sci. 2024. PMID: 38542484 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
