

Mice lacking MBNL1 and MBNL2 exhibit sudden cardiac death and molecular signatures recapitulating myotonic dystrophy
- PMID: 35567413
- PMCID: PMC9476621
- DOI: 10.1093/hmg/ddac108
Mice lacking MBNL1 and MBNL2 exhibit sudden cardiac death and molecular signatures recapitulating myotonic dystrophy
Abstract
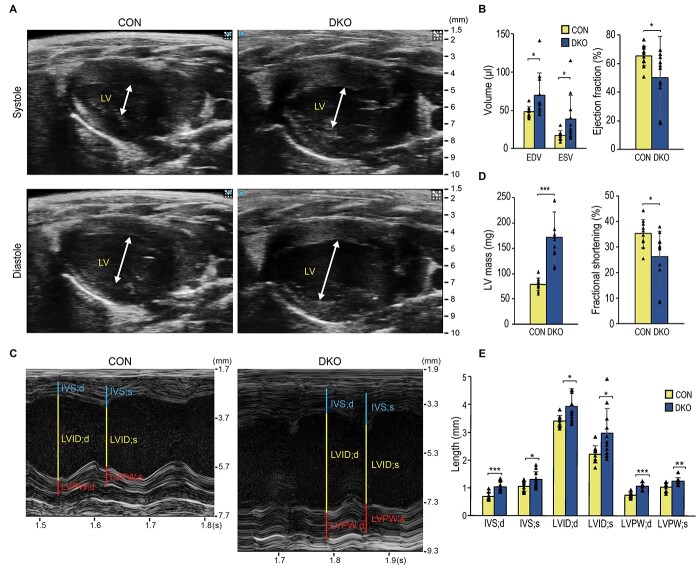
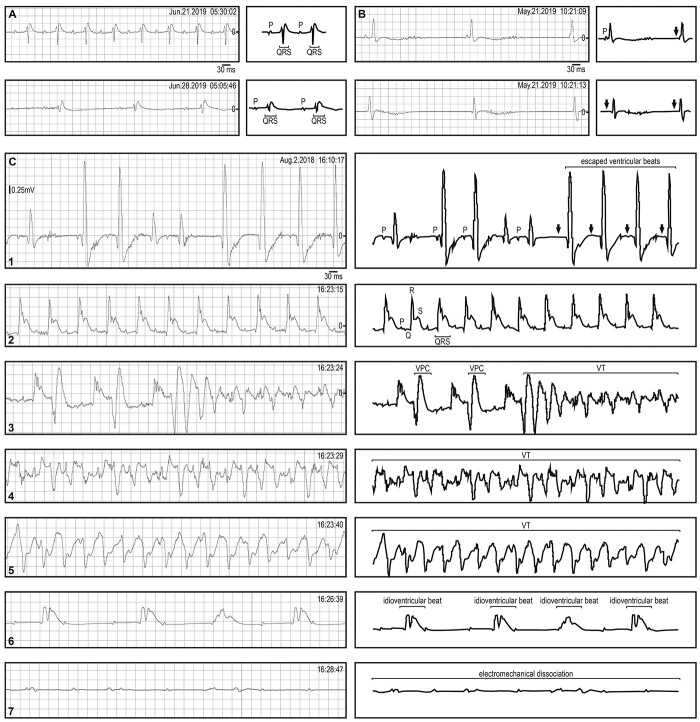
Myotonic dystrophy (DM) is caused by expansions of C(C)TG repeats in the non-coding regions of the DMPK and CNBP genes, and DM patients often suffer from sudden cardiac death due to lethal conduction block or arrhythmia. Specific molecular changes that underlie DM cardiac pathology have been linked to repeat-associated depletion of Muscleblind-like (MBNL) 1 and 2 proteins and upregulation of CUGBP, Elav-like family member 1 (CELF1). Hypothesis solely targeting MBNL1 or CELF1 pathways that could address all the consequences of repeat expansion in heart remained inconclusive, particularly when the direct cause of mortality and results of transcriptome analyses remained undetermined in Mbnl compound knockout (KO) mice with cardiac phenotypes. Here, we develop Myh6-Cre double KO (DKO) (Mbnl1-/-; Mbnl2cond/cond; Myh6-Cre+/-) mice to eliminate Mbnl1/2 in cardiomyocytes and observe spontaneous lethal cardiac events under no anesthesia. RNA sequencing recapitulates DM heart spliceopathy and shows gene expression changes that were previously undescribed in DM heart studies. Notably, immunoblotting reveals a nearly 6-fold increase of Calsequestrin 1 and 50% reduction of epidermal growth factor proteins. Our findings demonstrate that complete ablation of MBNL1/2 in cardiomyocytes is essential for generating sudden death due to lethal cardiac rhythms and reveal potential mechanisms for DM heart pathogenesis.
© The Author(s) 2022. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
