

Resistance to DNA repair inhibitors in cancer
- PMID: 35567571
- PMCID: PMC9627783
- DOI: 10.1002/1878-0261.13224
Resistance to DNA repair inhibitors in cancer
Abstract
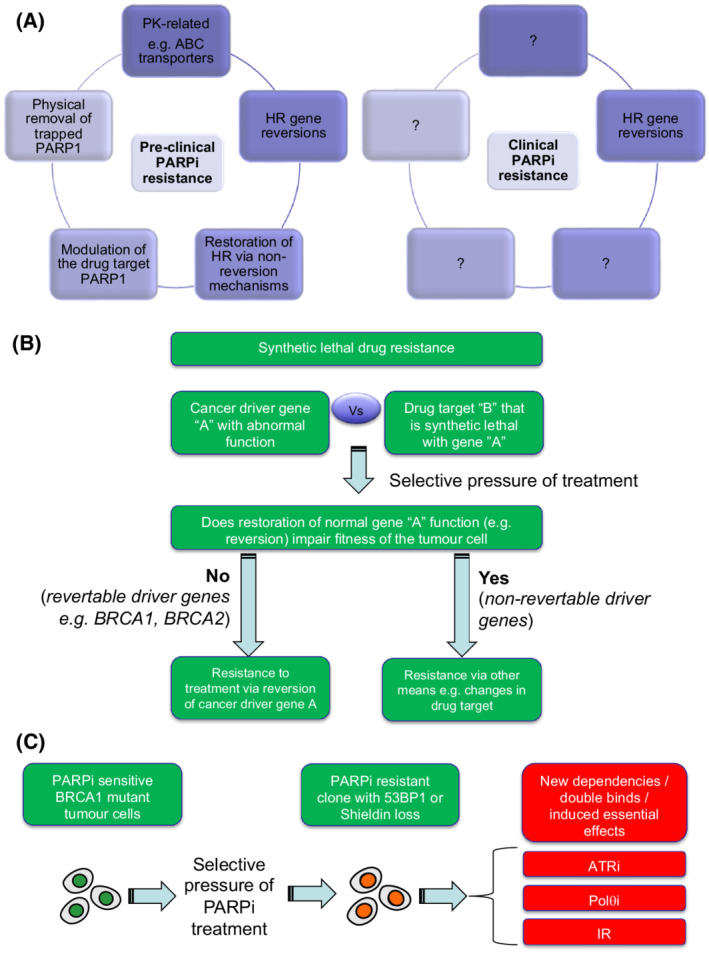
The DNA damage response (DDR) represents a complex network of proteins which detect and repair DNA damage, thereby maintaining the integrity of the genome and preventing the transmission of mutations and rearranged chromosomes to daughter cells. Faults in the DDR are a known driver and hallmark of cancer. Furthermore, inhibition of DDR enzymes can be used to treat the disease. This is exemplified by PARP inhibitors (PARPi) used to treat cancers with defects in the homologous recombination DDR pathway. A series of novel DDR targets are now also under pre-clinical or clinical investigation, including inhibitors of ATR kinase, WRN helicase or the DNA polymerase/helicase Polθ (Pol-Theta). Drug resistance is a common phenomenon that impairs the overall effectiveness of cancer treatments and there is already some understanding of how resistance to PARPi occurs. Here, we discuss how an understanding of PARPi resistance could inform how resistance to new drugs targeting the DDR emerges. We also discuss potential strategies that could limit the impact of these therapy resistance mechanisms in cancer.
Keywords: ATR; Cancer; DDR; PARP; PolQ; WRN.
© 2022 The Authors. Molecular Oncology published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
C.J.L. makes the following disclosures: receives and/or has received research funding from: AstraZeneca, Merck KGaA, Artios. Received consultancy, SAB membership or honoraria payments from: Syncona, Sun Pharma, Gerson Lehrman Group, Merck KGaA, Vertex, AstraZeneca, Tango, 3rd Rock, Ono Pharma, Artios, Abingworth, Tesselate, Dark Blue Therapeutics. Has stock in: Tango, Ovibio, Enedra Tx., Hysplex, Tesselate. C.J.L., D.Z. and S.J.P. are also named inventors on patents describing the use of DNA repair inhibitors and stand to gain from their development and use as part of the ICR ‘Rewards to Inventors’ scheme.
Figures
References
-
- Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46. 10.1158/2159-8290.CD-21-1059. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
