

New insight into dyslipidemia-induced cellular senescence in atherosclerosis
- PMID: 35569818
- PMCID: PMC9541442
- DOI: 10.1111/brv.12866
New insight into dyslipidemia-induced cellular senescence in atherosclerosis
Abstract
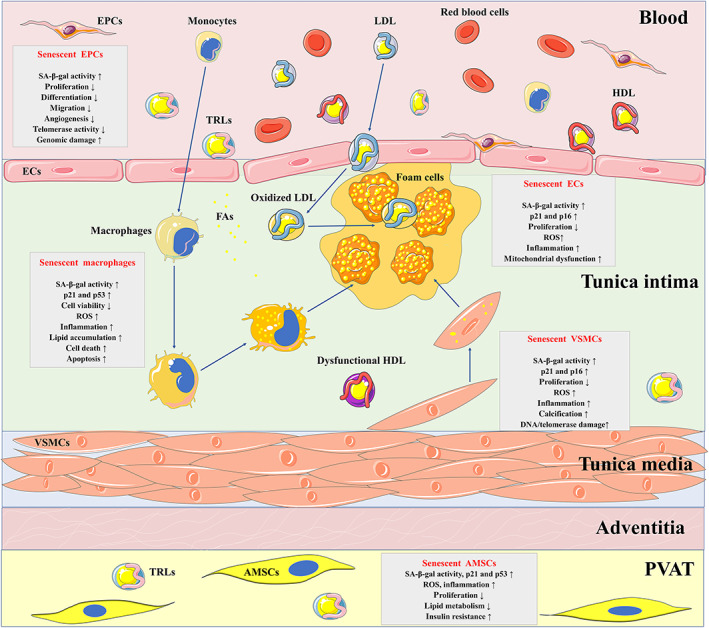
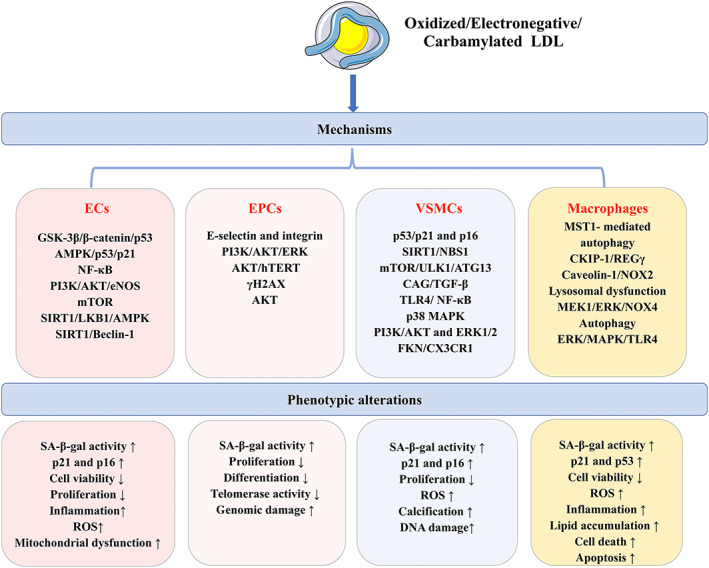
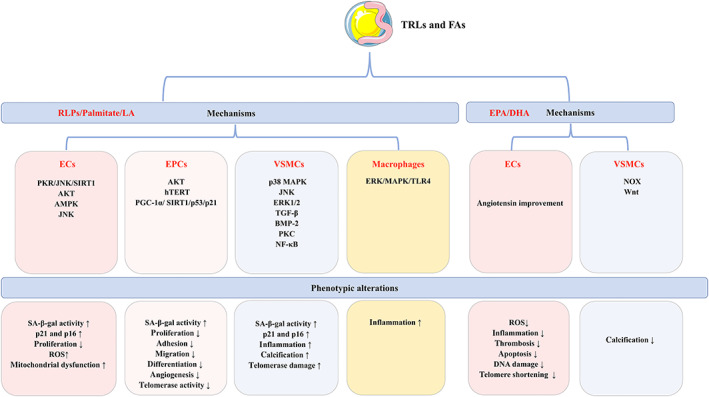
Atherosclerosis, characterized by lipid-rich plaques in the arterial wall, is an age-related disorder and a leading cause of mortality worldwide. However, the specific mechanisms remain complex. Recently, emerging evidence has demonstrated that senescence of various types of cells, such as endothelial cells (ECs), vascular smooth muscle cells (VSMCs), macrophages, endothelial progenitor cells (EPCs), and adipose-derived mesenchymal stem cells (AMSCs) contributes to atherosclerosis. Cellular senescence and atherosclerosis share various causative stimuli, in which dyslipidemia has attracted much attention. Dyslipidemia, mainly referred to elevated plasma levels of atherogenic lipids or lipoproteins, or functional impairment of anti-atherogenic lipids or lipoproteins, plays a pivotal role both in cellular senescence and atherosclerosis. In this review, we summarize the current evidence for dyslipidemia-induced cellular senescence during atherosclerosis, with a focus on low-density lipoprotein (LDL) and its modifications, hydrolysate of triglyceride-rich lipoproteins (TRLs), and high-density lipoprotein (HDL), respectively. Furthermore, we describe the underlying mechanisms linking dyslipidemia-induced cellular senescence and atherosclerosis. Finally, we discuss the senescence-related therapeutic strategies for atherosclerosis, with special attention given to the anti-atherosclerotic effects of promising geroprotectors as well as anti-senescence effects of current lipid-lowering drugs.
Keywords: adipose-derived mesenchymal stem cells; atherosclerosis; dyslipidemia; endothelial cells; macrophages; senescence; vascular smooth muscle cells.
© 2022 The Authors. Biological Reviews published by John Wiley & Sons Ltd on behalf of Cambridge Philosophical Society.
Figures
References
-
- Abe, Y. , Kawakami, A. , Osaka, M. , Uematsu, S. , Akira, S. , Shimokado, K. , Sacks, F. M. & Yoshida, M. (2010). Apolipoprotein CIII induces monocyte chemoattractant protein‐1 and interleukin 6 expression via toll‐like receptor 2 pathway in mouse adipocytes. Arteriosclerosis, Thrombosis, and Vascular Biology 30(11), 2242–2248. - PMC - PubMed
-
- Ai, D. , Jiang, H. , Westerterp, M. , Murphy, A. J. , Wang, M. , Ganda, A. , Abramowicz, S. , Welch, C. , Almazan, F. , Zhu, Y. , Miller, Y. I. & Tall, A. R. (2014). Disruption of mammalian target of rapamycin complex 1 in macrophages decreases chemokine gene expression and atherosclerosis. Circulation Research 114(10), 1576–1584. - PMC - PubMed
-
- Akhmedov, A. , Rozenberg, I. , Paneni, F. , Camici, G. G. , Shi, Y. , Doerries, C. , Sledzinska, A. , Mocharla, P. , Breitenstein, A. , Lohmann, C. , Stein, S. , von Lukowicz, T. , Kurrer, M. O. , Boren, J. , Becher, B. , et al. (2014). Endothelial overexpression of LOX‐1 increases plaque formation and promotes atherosclerosis in vivo. European Heart Journal 35(40), 2839–2848. - PubMed
-
- Akhmedov, A. , Camici, G. G. , Reiner, M. F. , Bonetti, N. R. , Costantino, S. , Holy, E. W. , Spescha, R. D. , Stivala, S. , Schaub Clerigue, A. , Speer, T. , Breitenstein, A. , Manz, J. , Lohmann, C. , Paneni, F. , Beer, J. H. , et al. (2017). Endothelial LOX‐1 activation differentially regulates arterial thrombus formation depending on oxLDL levels: role of the Oct‐1/SIRT1 and ERK1/2 pathways. Cardiovascscular Research 113(5), 498–507. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
